当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Carbon–Hydrogen Activation in Zerovalent Bis(1,5-cyclooctadiene) Complexes of the First Row Transition Metals: A Theoretical Study
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2018-03-05 00:00:00 , DOI: 10.1021/acs.jpca.7b12435 Jia Hu 1 , Hao Feng 1 , Yaoming Xie 2 , R. Bruce King 1, 2 , Henry F. Schaefer 2
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2018-03-05 00:00:00 , DOI: 10.1021/acs.jpca.7b12435 Jia Hu 1 , Hao Feng 1 , Yaoming Xie 2 , R. Bruce King 1, 2 , Henry F. Schaefer 2
Affiliation
|
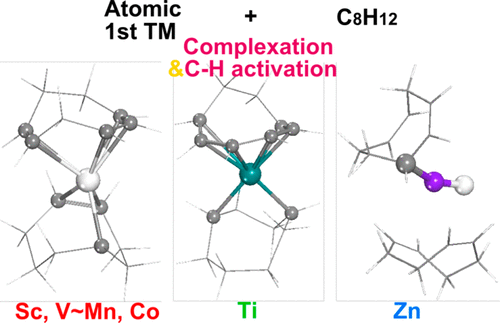
Stepwise interaction of first row transition metal atoms with 1,5-cyclooctadiene to give (C8H12)2M complexes is studied using the M06-L/DZP density functional method. The experimentally known (C8H12)2Ni is the thermodynamically most favorable complex, with a predicted geometry consistent with its experimental structure as determined by X-ray crystallography. The other transition metal atoms from scandium to zinc also interact exothermically with 1,5-cyclooctadiene to give (C8H12)2M derivatives, but these exhibit lower symmetry than the S4 symmetry exhibited by (C8H12)2Ni. Carbon–hydrogen activation of CH2 groups in a C8H12 ligand is predicted for most systems. Thus, conversion of (η2,2-C8H12)2M to (η3,2-C8H11)(η2,1-C8H13)M, through a hydride intermediate (η3,2-C8H11)(η2,2-C8H12)MH, is predicted for scandium, vanadium, chromium, manganese, and cobalt. For titanium with a low-lying empty orbital, further C–H activation through a hydride intermediate (η6-C8H10)(η2,1-C8H13)TiH is predicted, leading ultimately to (η6-C8H10)(η1,1-C8H14)Ti, in which the hexahapto η6-C8H10 ligand is shown by NICS to be aromatic. These two C–H activation processes on a titanium center represent the dehydrogenation of 1,5-cyclooctadiene to 1,3,5-cyclooctatriene with the second 1,5-cyclooctadiene ligand as the hydrogen acceptor. For zinc C–H activation terminates at (η1-C8H11)(C8H12)ZnH, which has a C–Zn–H three-center bond. No energetically favorable C–H activation processes are predicted for the iron, nickel, and copper (η2,2-C8H12)2M derivatives.
中文翻译:

第一排过渡金属的零价双(1,5-环辛二烯)配合物中的碳氢活化:理论研究
使用M06-L / DZP密度泛函方法研究了第一行过渡金属原子与1,5-环辛二烯的逐步相互作用以生成(C 8 H 12)2 M配合物。实验上已知的(C 8 H 12)2 Ni是热力学上最有利的配合物,其预测几何形状与其通过X射线晶体学测定的实验结构一致。从钪其它过渡金属原子也锌相互作用放热与1,5-环辛二烯,得到(C 8 H ^ 12)2剂M的衍生物,但比这些对称表现出较低的小号4由(C显示出对称8 H 12)2 Ni。预测大多数系统中C 8 H 12配体中CH 2基团的碳氢活化作用。因此,(η转化2,2- -C 8 ħ 12)2 M至(η 3,2 -C 8 ħ 11)(η 2,1 -C 8 ħ 13)M,通过氢化物中间体(η 3, 2 -C 8 ħ 11)(η 2,2- -C 8 ħ 12MH,预计用于for,钒,铬,锰和钴。对于通过与低洼空轨道,进一步C-H活化钛的氢化物中间体(η 6 -C 8 ħ 10)(η 2,1 -C 8 ħ 13)TIH预测,最终导致(η 6 - ç 8 ħ 10)(η 1,1- -C 8 ħ 14)的Ti,其中hexahaptoη 6 -C 8 ħ 10NICS显示该配体是芳族的。这两个在钛中心的CH活化过程代表了1,5-环辛二烯以第二个1,5-环辛二烯配体作为氢受体的脱氢反应为1,3,5-环辛三烯。在锌C-H活化终止(η 1 -C 8 ħ 11)(C 8 H ^ 12)ZNH,其具有C-ZNH三中心键。没有能量上有利的C-H活化方法被预测为铁,镍和铜(η 2,2- -C 8 ħ 12)2剂M的衍生物。
更新日期:2018-03-05
中文翻译:
第一排过渡金属的零价双(1,5-环辛二烯)配合物中的碳氢活化:理论研究
使用M06-L / DZP密度泛函方法研究了第一行过渡金属原子与1,5-环辛二烯的逐步相互作用以生成(C 8 H 12)2 M配合物。实验上已知的(C 8 H 12)2 Ni是热力学上最有利的配合物,其预测几何形状与其通过X射线晶体学测定的实验结构一致。从钪其它过渡金属原子也锌相互作用放热与1,5-环辛二烯,得到(C 8 H ^ 12)2剂M的衍生物,但比这些对称表现出较低的小号4由(C显示出对称8 H 12)2 Ni。预测大多数系统中C 8 H 12配体中CH 2基团的碳氢活化作用。因此,(η转化2,2- -C 8 ħ 12)2 M至(η 3,2 -C 8 ħ 11)(η 2,1 -C 8 ħ 13)M,通过氢化物中间体(η 3, 2 -C 8 ħ 11)(η 2,2- -C 8 ħ 12MH,预计用于for,钒,铬,锰和钴。对于通过与低洼空轨道,进一步C-H活化钛的氢化物中间体(η 6 -C 8 ħ 10)(η 2,1 -C 8 ħ 13)TIH预测,最终导致(η 6 - ç 8 ħ 10)(η 1,1- -C 8 ħ 14)的Ti,其中hexahaptoη 6 -C 8 ħ 10NICS显示该配体是芳族的。这两个在钛中心的CH活化过程代表了1,5-环辛二烯以第二个1,5-环辛二烯配体作为氢受体的脱氢反应为1,3,5-环辛三烯。在锌C-H活化终止(η 1 -C 8 ħ 11)(C 8 H ^ 12)ZNH,其具有C-ZNH三中心键。没有能量上有利的C-H活化方法被预测为铁,镍和铜(η 2,2- -C 8 ħ 12)2剂M的衍生物。










































 京公网安备 11010802027423号
京公网安备 11010802027423号