当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Structural Dynamics of Carbon Dots in Water and N, N-Dimethylformamide Probed by All-Atom Molecular Dynamics Simulations.
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2018-03-09 , DOI: 10.1021/acs.jctc.7b01149 Markéta Paloncýová 1 , Michal Langer 1 , Michal Otyepka 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2018-03-09 , DOI: 10.1021/acs.jctc.7b01149 Markéta Paloncýová 1 , Michal Langer 1 , Michal Otyepka 1
Affiliation
|
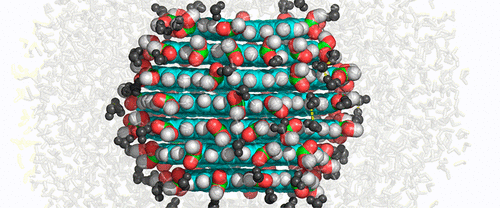
Carbon dots (CDs), one of the youngest members of the carbon nanostructure family, are now widely experimentally studied for their tunable fluorescence properties, bleaching resistance, and biocompatibility. Their interaction with biomolecular systems has also been explored experimentally. However, many atomistic details still remain unresolved. Molecular dynamics (MD) simulations enabling atomistic and femtosecond resolutions simultaneously are a well-established tool of computational chemistry which can provide useful insights into investigated systems. Here we present a full procedure for performing MD simulations of CDs. We developed a builder for generating CDs of a desired size and with various oxygen-containing surface functional groups. Further, we analyzed the behavior of various CDs differing in size, surface functional groups, and degrees of functionalization by MD simulations. These simulations showed that surface functionalized CDs are stable in a water environment through the formation of an extensive hydrogen bonding network. We also analyzed the internal dynamics of individual layers of CDs and evaluated the role of surface functional groups on CD stability. We observed that carboxyl groups interconnected the neighboring layers and decreased the rate of internal rotations. Further, we monitored changes in the CD shape caused by an excess of charged carboxyl groups or carbonyl groups. In addition to simulations in water, we analyzed the behavior of CDs in the organic solvent DMF, which decreased the stability of pure CDs but increased the level of interlayer hydrogen bonding. We believe that the developed protocol, builder, and parameters will facilitate future studies addressing various aspects of structural features of CDs and nanocomposites containing CDs.
中文翻译:

全原子分子动力学模拟探测水中碳点和N,N-二甲基甲酰胺的结构动力学。
碳点(CD)是碳纳米结构家族中最年轻的成员之一,目前因其可调的荧光特性,耐漂白性和生物相容性而受到广泛的实验研究。它们与生物分子系统的相互作用也已经通过实验进行了探索。但是,许多原子细节仍未解决。同时实现原子分辨率和飞秒分辨率的分子动力学(MD)模拟是一种成熟的计算化学工具,可以为研究系统提供有用的见识。在这里,我们介绍了执行CD的MD模拟的完整过程。我们开发了一种生成器,用于生成所需大小的CD和各种含氧表面官能团。此外,我们分析了各种CD的行为,这些CD的大小,表面官能团不同,和通过MD模拟的功能化程度。这些模拟表明,通过形成广泛的氢键网络,表面官能化的CD在水环境中是稳定的。我们还分析了CD各个层的内部动力学,并评估了表面官能团对CD稳定性的作用。我们观察到羧基使相邻层相互连接并降低了内部旋转的速率。此外,我们监测了由过量带电的羧基或羰基引起的CD形状的变化。除了在水中进行模拟外,我们还分析了CD在有机溶剂DMF中的行为,该行为降低了纯CD的稳定性,但增加了层间氢键的水平。我们认为,已开发的协议,构建器,
更新日期:2018-03-02
中文翻译:
全原子分子动力学模拟探测水中碳点和N,N-二甲基甲酰胺的结构动力学。
碳点(CD)是碳纳米结构家族中最年轻的成员之一,目前因其可调的荧光特性,耐漂白性和生物相容性而受到广泛的实验研究。它们与生物分子系统的相互作用也已经通过实验进行了探索。但是,许多原子细节仍未解决。同时实现原子分辨率和飞秒分辨率的分子动力学(MD)模拟是一种成熟的计算化学工具,可以为研究系统提供有用的见识。在这里,我们介绍了执行CD的MD模拟的完整过程。我们开发了一种生成器,用于生成所需大小的CD和各种含氧表面官能团。此外,我们分析了各种CD的行为,这些CD的大小,表面官能团不同,和通过MD模拟的功能化程度。这些模拟表明,通过形成广泛的氢键网络,表面官能化的CD在水环境中是稳定的。我们还分析了CD各个层的内部动力学,并评估了表面官能团对CD稳定性的作用。我们观察到羧基使相邻层相互连接并降低了内部旋转的速率。此外,我们监测了由过量带电的羧基或羰基引起的CD形状的变化。除了在水中进行模拟外,我们还分析了CD在有机溶剂DMF中的行为,该行为降低了纯CD的稳定性,但增加了层间氢键的水平。我们认为,已开发的协议,构建器,










































 京公网安备 11010802027423号
京公网安备 11010802027423号