当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Steered molecular dynamics simulations for uncovering the molecular mechanisms of drug dissociation and for drug screening: A test on the focal adhesion kinase
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2018-03-02 , DOI: 10.1002/jcc.25201 Chung F. Wong 1
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2018-03-02 , DOI: 10.1002/jcc.25201 Chung F. Wong 1
Affiliation
|
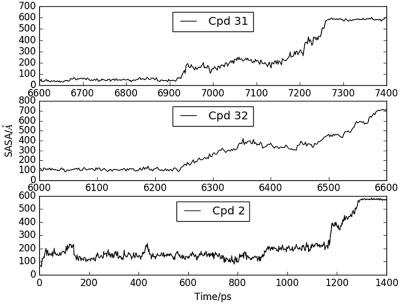
Drug‐binding kinetics could play important roles in determining the efficacy of drugs and has caught the attention of more drug designers. Using the dissociation of 1H‐pyrrolo[2,3‐b]‐pyridines from the focal adhesion kinase as an example, this work finds that steered molecular dynamics simulations could help screen compounds with long‐residence times. It also reveals a two‐step mechanism of ligand dissociation resembling the release of ADP from protein kinase A reported earlier. A phenyl group attaching to the pyrrole prolongs residence time by creating a large activation barrier for transition from the bound to the intermediate state when it becomes exposed to the solvent. Principal component analysis shows that ligand dissociation does not couple with large‐scale collective motions of the protein involving many of its amino acids. Rather, a small subset of amino acids dominates. Some of these amino acids do not contact the ligands directly along the dissociation pathways and could exert long‐range allosteric effects. © 2018 Wiley Periodicals, Inc.
中文翻译:

用于揭示药物解离和药物筛选的分子机制的导向分子动力学模拟:粘着斑激酶的测试
药物结合动力学可以在确定药物疗效方面发挥重要作用,并引起了更多药物设计者的关注。以 1H-吡咯并[2,3-b]-吡啶与粘着斑激酶的解离为例,这项工作发现,受控分子动力学模拟可以帮助筛选具有长驻留时间的化合物。它还揭示了配体解离的两步机制,类似于早先报道的蛋白激酶 A 释放 ADP。当它暴露于溶剂时,连接到吡咯的苯基通过产生大的活化势垒来延长停留时间,以从结合状态转变为中间状态。主成分分析表明,配体解离与涉及其许多氨基酸的蛋白质的大规模集体运动无关。相当,一小部分氨基酸占主导地位。这些氨基酸中的一些不沿着解离途径直接接触配体,并且可以发挥长程变构效应。© 2018 Wiley Periodicals, Inc.
更新日期:2018-03-02
中文翻译:
用于揭示药物解离和药物筛选的分子机制的导向分子动力学模拟:粘着斑激酶的测试
药物结合动力学可以在确定药物疗效方面发挥重要作用,并引起了更多药物设计者的关注。以 1H-吡咯并[2,3-b]-吡啶与粘着斑激酶的解离为例,这项工作发现,受控分子动力学模拟可以帮助筛选具有长驻留时间的化合物。它还揭示了配体解离的两步机制,类似于早先报道的蛋白激酶 A 释放 ADP。当它暴露于溶剂时,连接到吡咯的苯基通过产生大的活化势垒来延长停留时间,以从结合状态转变为中间状态。主成分分析表明,配体解离与涉及其许多氨基酸的蛋白质的大规模集体运动无关。相当,一小部分氨基酸占主导地位。这些氨基酸中的一些不沿着解离途径直接接触配体,并且可以发挥长程变构效应。© 2018 Wiley Periodicals, Inc.










































 京公网安备 11010802027423号
京公网安备 11010802027423号