当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Molecular-Simulation-Driven Fragment Screening for the Discovery of New CXCL12 Inhibitors
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2018-02-26 00:00:00 , DOI: 10.1021/acs.jcim.7b00625 Gerard Martinez-Rosell 1 , Matt J. Harvey 2 , Gianni De Fabritiis 1, 3
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2018-02-26 00:00:00 , DOI: 10.1021/acs.jcim.7b00625 Gerard Martinez-Rosell 1 , Matt J. Harvey 2 , Gianni De Fabritiis 1, 3
Affiliation
|
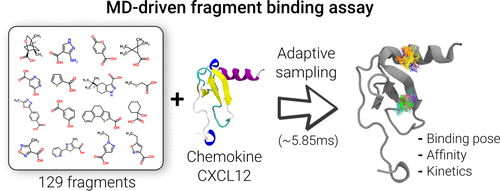
Fragment-based drug discovery (FBDD) has become a mainstream approach in drug design because it allows the reduction of the chemical space and screening libraries while identifying fragments with high protein–ligand efficiency interactions that can later be grown into drug-like leads. In this work, we leverage high-throughput molecular dynamics (MD) simulations to screen a library of 129 fragments for a total of 5.85 ms against the CXCL12 monomer, a chemokine involved in inflammation and diseases such as cancer. Our in silico binding assay was able to recover binding poses, affinities, and kinetics for the selected library and was able to predict 8 mM-affinity fragments with ligand efficiencies higher than 0.3. All of the fragment hits present a similar chemical structure, with a hydrophobic core and a positively charged group, and bind to either sY7 or H1S68 pockets, where they share pharmacophoric properties with experimentally resolved natural binders. This work presents a large-scale screening assay using an exclusive combination of thousands of short MD adaptive simulations analyzed with a Markov state model (MSM) framework.
中文翻译:

分子模拟驱动片段筛选的新型CXCL12抑制剂的发现。
基于片段的药物发现(FBDD)已成为药物设计中的主流方法,因为它可以减少化学空间并筛选文库,同时鉴定具有高蛋白-配体效率相互作用的片段,这些片段随后可以长成药物样的前导。在这项工作中,我们利用高通量分子动力学(MD)模拟来筛选针对CXCL12单体(涉及发炎和癌症等趋化因子)的129个片段的库,总共耗时5.85毫秒。我们的计算机结合分析能够恢复所选文库的结合姿势,亲和力和动力学,并能够预测8 mM亲和力片段,配体效率高于0.3。所有片段点击具有类似的化学结构,具有疏水核心和带正电荷的基团,并结合到sY7或H1S68口袋中,在口袋中它们与通过实验解决的天然粘合剂具有药理学特性。这项工作提出了一种大规模筛选分析方法,该方法使用了数千个简短的MD自适应模拟的独家组合,并通过马尔可夫状态模型(MSM)框架进行了分析。
更新日期:2018-02-26
中文翻译:
分子模拟驱动片段筛选的新型CXCL12抑制剂的发现。
基于片段的药物发现(FBDD)已成为药物设计中的主流方法,因为它可以减少化学空间并筛选文库,同时鉴定具有高蛋白-配体效率相互作用的片段,这些片段随后可以长成药物样的前导。在这项工作中,我们利用高通量分子动力学(MD)模拟来筛选针对CXCL12单体(涉及发炎和癌症等趋化因子)的129个片段的库,总共耗时5.85毫秒。我们的计算机结合分析能够恢复所选文库的结合姿势,亲和力和动力学,并能够预测8 mM亲和力片段,配体效率高于0.3。所有片段点击具有类似的化学结构,具有疏水核心和带正电荷的基团,并结合到sY7或H1S68口袋中,在口袋中它们与通过实验解决的天然粘合剂具有药理学特性。这项工作提出了一种大规模筛选分析方法,该方法使用了数千个简短的MD自适应模拟的独家组合,并通过马尔可夫状态模型(MSM)框架进行了分析。








































 京公网安备 11010802027423号
京公网安备 11010802027423号