当前位置:
X-MOL 学术
›
Faraday Discuss.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Crystal structure prediction of flexible pharmaceutical-like molecules: density functional tight-binding as an intermediate optimisation method and for free energy estimation
Faraday Discussions ( IF 3.4 ) Pub Date : 2018-02-27 , DOI: 10.1039/c8fd00010g Luca Iuzzolino 1, 2, 3, 4 , Patrick McCabe 4, 5, 6 , Sarah L. Price 1, 2, 3, 4 , Jan Gerit Brandenburg 1, 2, 3, 4
Faraday Discussions ( IF 3.4 ) Pub Date : 2018-02-27 , DOI: 10.1039/c8fd00010g Luca Iuzzolino 1, 2, 3, 4 , Patrick McCabe 4, 5, 6 , Sarah L. Price 1, 2, 3, 4 , Jan Gerit Brandenburg 1, 2, 3, 4
Affiliation
|
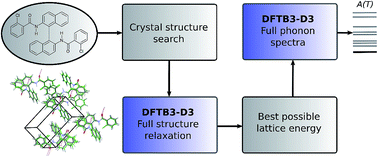
Successful methodologies for theoretical crystal structure prediction (CSP) on flexible pharmaceutical-like organic molecules explore the lattice energy surface to find a set of plausible crystal structures. The initial search stages of CSP studies use relatively simple lattice energy approximations as hundreds of thousands of minima have to be considered. These generated crystal structures often have poor molecular geometries, as well as inaccurate lattice energy rankings, and performing reasonably accurate but computationally affordable optimisations of the crystal structures generated in a search would be highly desirable. Here, we seek to explore whether semi-empirical quantum-mechanical methods can perform this task. We employed the dispersion-corrected tight-binding Hamiltonian (DFTB3-D3) to relax all the inter- and intra-molecular degrees of freedom of several thousands of generated crystal structures of five pharmaceutical-like molecules, saving a large amount of computational effort compared to earlier studies. The computational cost scales better with molecular size and flexibility than other CSP methods, suggesting that it could be extended to even larger and more flexible molecules. On average, this optimisation improved the average reproduction of the eight experimental crystal structures (RMSD15) and experimental conformers (RMSD1) by 4% and 23%, respectively. The intermolecular interactions were then further optimised using distributed multipoles, derived from the molecular wave-functions, to accurately describe the electrostatic components of the intermolecular energies. In all cases, the experimental crystal structures are close to the top of the lattice energy ranking. Phonon calculations on some of the lowest energy structures were also performed with DFTB3-D3 methods to calculate the vibrational component of the Helmholtz free energy, providing further insights into the solid-state behaviour of the target molecules. We conclude that DFTB3-D3 is a cost-effective method for optimising flexible molecules, bridging the gap between the approximate methods used in CSP searches for generating crystal structures and more accurate methods required in the final energy ranking.
中文翻译:

柔性药物样分子的晶体结构预测:作为中间优化方法和自由能估计的密度泛函紧密结合
在类药物柔性有机分子上进行理论晶体结构预测(CSP)的成功方法论,探索了晶格能表面,从而找到了一组合理的晶体结构。CSP研究的初始搜索阶段使用相对简单的晶格能量近似,因为必须考虑成千上万的最小值。这些生成的晶体结构通常具有较差的分子几何形状以及不准确的晶格能量等级,并且非常需要对搜索中生成的晶体结构进行合理准确但在计算上可承受的优化。在这里,我们试图探索半经验量子力学方法是否可以执行此任务。我们采用了分散校正的紧密结合的哈密顿量(DFTB3-D3)来松弛五个药物样分子的数千个晶体结构的所有分子间和分子内自由度,与之相比,节省了大量的计算工作量到较早的研究。与其他CSP方法相比,在分子大小和灵活性方面,计算成本的伸缩性更好,这表明它可以扩展到更大,更灵活的分子。平均而言,这种优化改善了八个实验晶体结构(RMSD 与其他CSP方法相比,在分子大小和灵活性方面,计算成本的伸缩性更好,这表明它可以扩展到更大,更灵活的分子。平均而言,这种优化改善了八个实验晶体结构(RMSD 与其他CSP方法相比,在分子大小和灵活性方面,计算成本的伸缩性更好,这表明它可以扩展到更大,更灵活的分子。平均而言,这种优化改善了八个实验晶体结构(RMSD15)和实验整合者(RMSD 1)分别增加4%和23%。然后使用衍生自分子波函数的分布多极子进一步优化分子间的相互作用,以准确描述分子间能量的静电成分。在所有情况下,实验晶体结构都接近晶格能量等级的顶部。还使用DFTB3-D3方法对一些最低能级结构进行了声子计算,以计算亥姆霍兹自由能的振动分量,从而进一步了解了目标分子的固态行为。我们得出结论,DFTB3-D3是优化柔性分子的一种经济有效的方法,弥合了CSP搜索中用于生成晶体结构的近似方法与最终能量排名所需的更精确方法之间的差距。
更新日期:2018-10-26
中文翻译:
柔性药物样分子的晶体结构预测:作为中间优化方法和自由能估计的密度泛函紧密结合
在类药物柔性有机分子上进行理论晶体结构预测(CSP)的成功方法论,探索了晶格能表面,从而找到了一组合理的晶体结构。CSP研究的初始搜索阶段使用相对简单的晶格能量近似,因为必须考虑成千上万的最小值。这些生成的晶体结构通常具有较差的分子几何形状以及不准确的晶格能量等级,并且非常需要对搜索中生成的晶体结构进行合理准确但在计算上可承受的优化。在这里,我们试图探索半经验量子力学方法是否可以执行此任务。我们采用了分散校正的紧密结合的哈密顿量(DFTB3-D3)来松弛五个药物样分子的数千个晶体结构的所有分子间和分子内自由度,与之相比,节省了大量的计算工作量到较早的研究。与其他CSP方法相比,在分子大小和灵活性方面,计算成本的伸缩性更好,这表明它可以扩展到更大,更灵活的分子。平均而言,这种优化改善了八个实验晶体结构(RMSD 与其他CSP方法相比,在分子大小和灵活性方面,计算成本的伸缩性更好,这表明它可以扩展到更大,更灵活的分子。平均而言,这种优化改善了八个实验晶体结构(RMSD 与其他CSP方法相比,在分子大小和灵活性方面,计算成本的伸缩性更好,这表明它可以扩展到更大,更灵活的分子。平均而言,这种优化改善了八个实验晶体结构(RMSD15)和实验整合者(RMSD 1)分别增加4%和23%。然后使用衍生自分子波函数的分布多极子进一步优化分子间的相互作用,以准确描述分子间能量的静电成分。在所有情况下,实验晶体结构都接近晶格能量等级的顶部。还使用DFTB3-D3方法对一些最低能级结构进行了声子计算,以计算亥姆霍兹自由能的振动分量,从而进一步了解了目标分子的固态行为。我们得出结论,DFTB3-D3是优化柔性分子的一种经济有效的方法,弥合了CSP搜索中用于生成晶体结构的近似方法与最终能量排名所需的更精确方法之间的差距。


























 京公网安备 11010802027423号
京公网安备 11010802027423号