当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
New tricks for old dogs: improving the accuracy of biomolecular force fields by pair-specific corrections to non-bonded interactions†
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2018-02-26 00:00:00 , DOI: 10.1039/c7cp08185e Jejoong Yoo 1, 2, 3, 4, 5 , Aleksei Aksimentiev 1, 2, 3, 4, 5
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2018-02-26 00:00:00 , DOI: 10.1039/c7cp08185e Jejoong Yoo 1, 2, 3, 4, 5 , Aleksei Aksimentiev 1, 2, 3, 4, 5
Affiliation
|
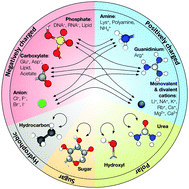
In contrast to ordinary polymers, the vast majority of biological macromolecules adopt highly ordered three-dimensional structures that define their functions. The key to folding of a biopolymer into a unique 3D structure or to an assembly of several biopolymers into a functional unit is a delicate balance between the attractive and repulsive forces that also makes such self-assembly reversible under physiological conditions. The all-atom molecular dynamics (MD) method has emerged as a powerful tool for studies of individual biomolecules and their functional assemblies, encompassing systems of ever increasing complexity. However, advances in parallel computing technology have outpaced the development of the underlying theoretical models—the molecular force fields, pushing the MD method into an untested territory. Recent tests of the MD method have found the most commonly used molecular force fields to be out of balance, overestimating attractive interactions between charged and hydrophobic groups, which can promote artificial aggregation in MD simulations of multi-component protein, nucleic acid, and lipid systems. One route towards improving the force fields is through the NBFIX corrections method, in which the intermolecular forces are calibrated against experimentally measured quantities such as osmotic pressure by making atom pair-specific adjustments to the non-bonded interactions. In this article, we review development of the NBFIX (Non-Bonded FIX) corrections to the AMBER and CHARMM force fields and discuss their implications for MD simulations of electrolyte solutions, dense DNA systems, Holliday junctions, protein folding, and lipid bilayer membranes.
中文翻译:

老年犬的新技巧:通过对非键相互作用的成对特定校正来提高生物分子力场的准确性†
与普通聚合物相反,绝大多数生物大分子采用高度有序的三维结构来定义其功能。将生物聚合物折叠成独特的3D结构或将几种生物聚合物组装成功能单元的关键是吸引力和排斥力之间的微妙平衡,这也使得这种自组装在生理条件下可逆。全原子分子动力学(MD)方法已成为研究单个生物分子及其功能组件的强大工具,涵盖了日益复杂的系统。但是,并行计算技术的发展已经超过了基础理论模型(分子力场)的发展速度,从而将MD方法推向了未经测试的领域。MD方法的最新测试发现最常用的分子力场失衡,高估了带电基团和疏水基团之间的吸引力,这可能会在多组分蛋白质,核酸和脂质系统的MD模拟中促进人工聚集。改善力场的一种途径是通过NBFIX校正方法,其中通过对非键合相互作用进行原子对特定的调节,根据实验测量的量(例如渗透压)校准分子间力。在本文中,我们回顾了针对AMBER和CHARMM力场的NBFIX(非键合FIX)校正的发展,并讨论了它们对电解质溶液,致密DNA系统,霍利迪结,蛋白质折叠,
更新日期:2018-02-26
中文翻译:
老年犬的新技巧:通过对非键相互作用的成对特定校正来提高生物分子力场的准确性†
与普通聚合物相反,绝大多数生物大分子采用高度有序的三维结构来定义其功能。将生物聚合物折叠成独特的3D结构或将几种生物聚合物组装成功能单元的关键是吸引力和排斥力之间的微妙平衡,这也使得这种自组装在生理条件下可逆。全原子分子动力学(MD)方法已成为研究单个生物分子及其功能组件的强大工具,涵盖了日益复杂的系统。但是,并行计算技术的发展已经超过了基础理论模型(分子力场)的发展速度,从而将MD方法推向了未经测试的领域。MD方法的最新测试发现最常用的分子力场失衡,高估了带电基团和疏水基团之间的吸引力,这可能会在多组分蛋白质,核酸和脂质系统的MD模拟中促进人工聚集。改善力场的一种途径是通过NBFIX校正方法,其中通过对非键合相互作用进行原子对特定的调节,根据实验测量的量(例如渗透压)校准分子间力。在本文中,我们回顾了针对AMBER和CHARMM力场的NBFIX(非键合FIX)校正的发展,并讨论了它们对电解质溶液,致密DNA系统,霍利迪结,蛋白质折叠,










































 京公网安备 11010802027423号
京公网安备 11010802027423号