当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Towards reliable references for electron paramagnetic resonance parameters based on quantum chemistry: the case of verdazyl radicals†
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2018-02-26 00:00:00 , DOI: 10.1039/c7cp05657e Anja Massolle 1, 2, 3, 4, 5 , Thomas Dresselhaus 1, 2, 3, 4, 5 , Steffen Eusterwiemann 1, 2, 3, 4 , Carsten Doerenkamp 2, 3, 4, 6, 7 , Hellmut Eckert 2, 3, 4, 6, 7 , Armido Studer 1, 2, 3, 4 , Johannes Neugebauer 1, 2, 3, 4, 5
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2018-02-26 00:00:00 , DOI: 10.1039/c7cp05657e Anja Massolle 1, 2, 3, 4, 5 , Thomas Dresselhaus 1, 2, 3, 4, 5 , Steffen Eusterwiemann 1, 2, 3, 4 , Carsten Doerenkamp 2, 3, 4, 6, 7 , Hellmut Eckert 2, 3, 4, 6, 7 , Armido Studer 1, 2, 3, 4 , Johannes Neugebauer 1, 2, 3, 4, 5
Affiliation
|
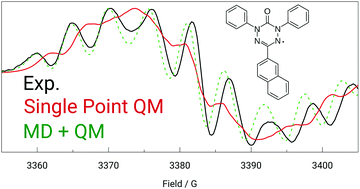
We present an efficient and accurate computational procedure to calculate properties measurable by EPR spectroscopy. We simulate a molecular dynamics (MD) trajectory by employing the quantum mechanically derived force field (QMDFF) [S. Grimme, J. Chem. Theory Comput., 2014, 10, 4497] and sample the trajectory at different time steps. For each snapshot EPR properties are calculated with a hybrid density functional theory (DFT) method. EPR spectra are simulated based on the averaged results. We applied the strategy to a number of previously published and novel verdazyl radicals, for which we recorded EPR spectra. The resulting simulated spectra are compatible with experiment already before employing an additional fitting step, in contrast to those from single point electronic-structure calculations. After the refinement, the experimental data are excellently reproduced, and the fitted EPR parameters do not deviate much from the calculated ones. This provides confidence in ascribing a direct physical meaning to the refined data in terms of experimental EPR parameters rather than merely considering them as mathematical fit parameters. We also find that couplings to hydrogen nuclei have a significant influence on the spectra of verdazyl radicals.
中文翻译:

寻求基于量子化学的电子顺磁共振参数的可靠参考:维达唑基的情况†
我们提出了一种高效,准确的计算程序来计算可通过EPR光谱测量的特性。我们通过利用量子机械推导的力场(QMDFF)[S。格里姆,化学杂志。理论计算。,2014年,10,4497],并在不同的时间步长对轨迹进行采样。对于每个快照,均使用混合密度泛函理论(DFT)方法计算EPR属性。根据平均结果模拟EPR光谱。我们将该策略应用于许多先前发布的和新颖的verdazyl自由基,为此我们记录了EPR光谱。与单点电子结构计算得出的结果相反,所得到的模拟光谱与采用附加拟合步骤之前的实验已经兼容。改进后,可以很好地复制实验数据,并且拟合的EPR参数与计算的参数相差不大。这提供了根据实验EPR参数而不是仅仅将其视为数学拟合参数而将精炼数据具有直接物理意义的信心。我们还发现与氢核的偶联对Verdazyl自由基的光谱有重要影响。
更新日期:2018-02-26
中文翻译:
寻求基于量子化学的电子顺磁共振参数的可靠参考:维达唑基的情况†
我们提出了一种高效,准确的计算程序来计算可通过EPR光谱测量的特性。我们通过利用量子机械推导的力场(QMDFF)[S。格里姆,化学杂志。理论计算。,2014年,10,4497],并在不同的时间步长对轨迹进行采样。对于每个快照,均使用混合密度泛函理论(DFT)方法计算EPR属性。根据平均结果模拟EPR光谱。我们将该策略应用于许多先前发布的和新颖的verdazyl自由基,为此我们记录了EPR光谱。与单点电子结构计算得出的结果相反,所得到的模拟光谱与采用附加拟合步骤之前的实验已经兼容。改进后,可以很好地复制实验数据,并且拟合的EPR参数与计算的参数相差不大。这提供了根据实验EPR参数而不是仅仅将其视为数学拟合参数而将精炼数据具有直接物理意义的信心。我们还发现与氢核的偶联对Verdazyl自由基的光谱有重要影响。








































 京公网安备 11010802027423号
京公网安备 11010802027423号