Journal of Catalysis ( IF 6.5 ) Pub Date : 2018-02-22 , DOI: 10.1016/j.jcat.2018.01.035 Thomas Reichenbach , Krishnakanta Mondal , Marc Jäger , Thomas Vent-Schmidt , Daniel Himmel , Valentin Dybbert , Albert Bruix , Ingo Krossing , Michael Walter , Michael Moseler
|
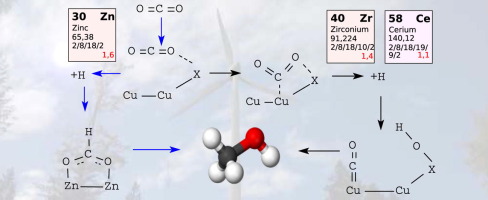
Methanol formation from CO2 and molecular hydrogen on ZnO/Cu catalysts is studied by gradient corrected density functional theory. The catalytically active region is modeled as a minimum size inverse catalyst represented by ZnXOY(H) clusters of different size and a ZnO nano-ribbon on an extended Cu(1 1 1) surface. These systems are chosen as a representative of thermodynamically stable catalyst structures under typical reaction conditions. Comparison to a high level wave function method reveals that density functional theory systematically underestimates reaction barriers, but nevertheless conserves their energetic ordering. In contrast to other metal-supported oxides like ceria and zirconia, the reaction proceeds through the formation of formate on ZnOX/Cu, thus avoiding the CO intermediate. The difference between the oxides is attributed to variance in the initial activation of CO2. The energetics of the formate reaction pathway is insensitive to the exact environment of undercoordinated Zn active sites, which points to a general mechanism for Cu-Zn based catalysts.
中文翻译:
反向ZnO / Cu催化剂CO 2加氢机理的从头算研究
利用梯度校正密度泛函理论研究了在ZnO / Cu催化剂上由CO 2和分子氢形成甲醇的过程。催化活性区域建模为最小尺寸的逆催化剂,该催化剂由不同尺寸的Zn X O Y(H)簇和扩展的Cu(1 1 1)表面上的ZnO纳米带表示。选择这些系统作为典型反应条件下热力学稳定催化剂结构的代表。与高水平波函数方法的比较表明,密度泛函理论系统地低估了反应势垒,但仍保留了它们的能量有序。与其他金属负载的氧化物(如二氧化铈和氧化锆)相反,该反应通过在ZnO上形成甲酸盐来进行X / Cu,因此避免了CO中间体。氧化物之间的差异归因于CO 2的初始活化中的变化。甲酸反应途径的能量学对未配位的Zn活性位点的确切环境不敏感,这表明Cu-Zn基催化剂的一般机理。










































 京公网安备 11010802027423号
京公网安备 11010802027423号