当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Cis-to-Trans Isomerization of Azobenzene Derivatives Studied with Transition Path Sampling and Quantum Mechanical/Molecular Mechanical Molecular Dynamics
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2018-02-21 00:00:00 , DOI: 10.1021/acs.jctc.7b01120 Anja Muždalo 1 , Peter Saalfrank 2 , Jocelyne Vreede 3 , Mark Santer 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2018-02-21 00:00:00 , DOI: 10.1021/acs.jctc.7b01120 Anja Muždalo 1 , Peter Saalfrank 2 , Jocelyne Vreede 3 , Mark Santer 1
Affiliation
|
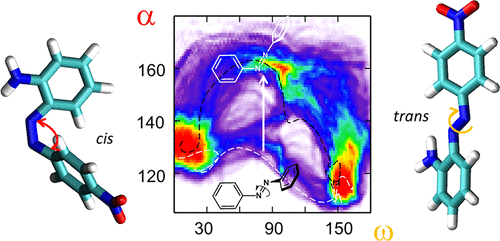
Azobenzene-based molecular photoswitches are becoming increasingly important for the development of photoresponsive, functional soft-matter material systems. Upon illumination with light, fast interconversion between a more stable trans and a metastable cis configuration can be established resulting in pronounced changes in conformation, dipole moment or hydrophobicity. A rational design of functional photosensitive molecules with embedded azo moieties requires a thorough understanding of isomerization mechanisms and rates, especially the thermally activated relaxation. For small azo derivatives considered in the gas phase or simple solvents, Eyring’s classical transition state theory (TST) approach yields useful predictions for trends in activation energies or corresponding half-life times of the cis isomer. However, TST or improved theories cannot easily be applied when the azo moiety is part of a larger molecular complex or embedded into a heterogeneous environment, where a multitude of possible reaction pathways may exist. In these cases, only the sampling of an ensemble of dynamic reactive trajectories (transition path sampling, TPS) with explicit models of the environment may reveal the nature of the processes involved. In the present work we show how a TPS approach can conveniently be implemented for the phenomenon of relaxation–isomerization of azobenzenes starting with the simple examples of pure azobenzene and a push–pull derivative immersed in a polar (DMSO) and apolar (toluene) solvent. The latter are represented explicitly at a molecular mechanical (MM) and the azo moiety at a quantum mechanical (QM) level. We demonstrate for the push–pull azobenzene that path sampling in combination with the chosen QM/MM scheme produces the expected change in isomerization pathway from inversion to rotation in going from a low to a high permittivity (explicit) solvent model. We discuss the potential of the simulation procedure presented for comparative calculation of reaction rates and an improved understanding of activated states.
中文翻译:

顺至-反式偶氮苯衍生物的异构化与转型路径取样和量子力学/分子力学分子动力学研究了
基于偶氮苯的分子光电开关对于光响应性功能性软物质材料系统的开发变得越来越重要。在光照下,更稳定的反式和亚稳态的顺式之间可以快速相互转换可以建立构型,导致构象,偶极矩或疏水性发生明显变化。具有嵌入的偶氮部分的功能性光敏分子的合理设计需要对异构化机理和速率,特别是热活化弛豫的透彻理解。对于气相中考虑的小型偶氮衍生物或简单溶剂,艾林的经典过渡态理论(TST)方法可得出有用的预测活化能或相应顺式半衰期的趋势的预测异构体。但是,当偶氮部分是较大分子复合物的一部分或嵌入异质环境(其中可能存在许多可能的反应路径)时,TST或改进的理论就不容易应用。在这些情况下,只有动态反应性轨迹的整体采样(过渡路径采样,TPS)和明确的环境模型才能揭示所涉及过程的性质。在当前的工作中,我们展示了如何从简单的纯偶氮苯和浸入极性(DMSO)和非极性(甲苯)溶剂中的推挽衍生物的简单示例开始,针对偶氮苯的弛豫-异构化现象方便地实施TPS方法。后者以分子力学(MM)的形式明确表示,而偶氮部分以量子力学(QM)的水平表示。对于推挽式偶氮苯,我们证明了路径采样与所选的QM / MM方案相结合,可实现从低介电常数到高介电常数(显式)溶剂模型的异构化途径从反转到旋转的预期变化。我们讨论了用于比较计算反应速率和提高对活化态的理解的模拟程序的潜力。
更新日期:2018-02-21
中文翻译:
顺至-反式偶氮苯衍生物的异构化与转型路径取样和量子力学/分子力学分子动力学研究了
基于偶氮苯的分子光电开关对于光响应性功能性软物质材料系统的开发变得越来越重要。在光照下,更稳定的反式和亚稳态的顺式之间可以快速相互转换可以建立构型,导致构象,偶极矩或疏水性发生明显变化。具有嵌入的偶氮部分的功能性光敏分子的合理设计需要对异构化机理和速率,特别是热活化弛豫的透彻理解。对于气相中考虑的小型偶氮衍生物或简单溶剂,艾林的经典过渡态理论(TST)方法可得出有用的预测活化能或相应顺式半衰期的趋势的预测异构体。但是,当偶氮部分是较大分子复合物的一部分或嵌入异质环境(其中可能存在许多可能的反应路径)时,TST或改进的理论就不容易应用。在这些情况下,只有动态反应性轨迹的整体采样(过渡路径采样,TPS)和明确的环境模型才能揭示所涉及过程的性质。在当前的工作中,我们展示了如何从简单的纯偶氮苯和浸入极性(DMSO)和非极性(甲苯)溶剂中的推挽衍生物的简单示例开始,针对偶氮苯的弛豫-异构化现象方便地实施TPS方法。后者以分子力学(MM)的形式明确表示,而偶氮部分以量子力学(QM)的水平表示。对于推挽式偶氮苯,我们证明了路径采样与所选的QM / MM方案相结合,可实现从低介电常数到高介电常数(显式)溶剂模型的异构化途径从反转到旋转的预期变化。我们讨论了用于比较计算反应速率和提高对活化态的理解的模拟程序的潜力。










































 京公网安备 11010802027423号
京公网安备 11010802027423号