当前位置:
X-MOL 学术
›
J. Phys. Chem. B
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Transferable Coarse-Grained Models of Liquid–Liquid Equilibrium Using Local Density Potentials Optimized with the Relative Entropy
The Journal of Physical Chemistry B ( IF 2.8 ) Pub Date : 2018-02-21 00:00:00 , DOI: 10.1021/acs.jpcb.7b12446 Tanmoy Sanyal 1 , M. Scott Shell 1
The Journal of Physical Chemistry B ( IF 2.8 ) Pub Date : 2018-02-21 00:00:00 , DOI: 10.1021/acs.jpcb.7b12446 Tanmoy Sanyal 1 , M. Scott Shell 1
Affiliation
|
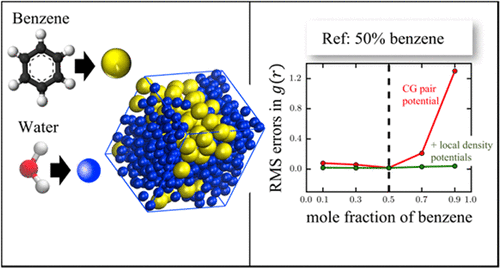
Bottom-up coarse-grained (CG) models are now regularly pursued to enable large length and time scale molecular simulations of complex, often macromolecular systems. However, predicting fluid phase equilibria using such models remains fundamentally challenging. A major problem stems from the typically low transferability of CG models beyond the densities and/or compositions at which they are parametrized, which is necessary if they are to describe distinct structural and thermodynamic properties unique to each phase. CG model transferability is compounded by the representation of the inherently multibody coarse interactions using pair potentials that neglect higher order effects. Here, we propose to construct transferable single site CG models of liquid mixtures by supplementing traditional CG pair interactions with local density potentials, which constitute a computationally inexpensive mean-field approach to describe many-body effects, in that site energies are modulated by the local solution environment. To illustrate the approach, we use intra- and interspecies local density potentials to develop CG models of benzene–water solutions that show impressive transferability in structural metrics (pair correlation functions, density profiles) throughout composition space, in contrast to pair-only CG representations. While further refinement may be necessary to represent more complex thermodynamic properties, like the liquid–liquid interfacial tension, the generality and improvement offered by the local density approach are highly encouraging for enabling complex phase equilibrium modeling using CG models.
中文翻译:

利用相对熵优化的局部密度势的液体-液体平衡的粗粒度可转移模型
现在,经常采用自下而上的粗粒度(CG)模型,以实现复杂的,通常是大分子系统的长时标分子模拟。但是,使用此类模型预测液相平衡仍然从根本上具有挑战性。一个主要问题是由于CG模型的典型低传递性超出了参数化所用的密度和/或组成,如果它们要描述每个相特有的独特结构和热力学性质,则这是必需的。CG模型的可传递性通过使用对电位忽略较高阶效应的固有多体粗糙相互作用的表示而变得更加复杂。在这里,我们建议通过补充具有局部密度势的传统CG对相互作用来构建液体混合物的可转移单点CG模型,它们构成了一种计算上不昂贵的均值场方法来描述多体效应,因为现场能量是由局部解环境调制的。为了说明这种方法,我们使用种间和种间局部密度势来开发苯-水溶液的CG模型,与仅成对的CG表示法相比,该模型在整个成分空间的结构度量(对相关函数,密度分布图)中显示出令人印象深刻的可转移性。尽管可能需要进一步细化以表示更复杂的热力学性质,例如液-液界面张力,但局部密度方法提供的一般性和改进方法对于使用CG模型进行复杂的相平衡建模非常令人鼓舞。
更新日期:2018-02-21
中文翻译:
利用相对熵优化的局部密度势的液体-液体平衡的粗粒度可转移模型
现在,经常采用自下而上的粗粒度(CG)模型,以实现复杂的,通常是大分子系统的长时标分子模拟。但是,使用此类模型预测液相平衡仍然从根本上具有挑战性。一个主要问题是由于CG模型的典型低传递性超出了参数化所用的密度和/或组成,如果它们要描述每个相特有的独特结构和热力学性质,则这是必需的。CG模型的可传递性通过使用对电位忽略较高阶效应的固有多体粗糙相互作用的表示而变得更加复杂。在这里,我们建议通过补充具有局部密度势的传统CG对相互作用来构建液体混合物的可转移单点CG模型,它们构成了一种计算上不昂贵的均值场方法来描述多体效应,因为现场能量是由局部解环境调制的。为了说明这种方法,我们使用种间和种间局部密度势来开发苯-水溶液的CG模型,与仅成对的CG表示法相比,该模型在整个成分空间的结构度量(对相关函数,密度分布图)中显示出令人印象深刻的可转移性。尽管可能需要进一步细化以表示更复杂的热力学性质,例如液-液界面张力,但局部密度方法提供的一般性和改进方法对于使用CG模型进行复杂的相平衡建模非常令人鼓舞。










































 京公网安备 11010802027423号
京公网安备 11010802027423号