当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Physisorption of H2 on Fullerenes and the Solvation of C60 by Hydrogen Clusters at Finite Temperature: A Theoretical Assessment
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2018-02-16 00:00:00 , DOI: 10.1021/acs.jpca.8b00163 F. Calvo 1 , E. Yurtsever 2 , A. Tekin 3
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2018-02-16 00:00:00 , DOI: 10.1021/acs.jpca.8b00163 F. Calvo 1 , E. Yurtsever 2 , A. Tekin 3
Affiliation
|
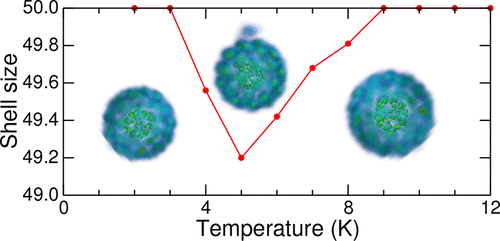
The interaction between hydrogen and carbonaceous nanostructures is of fundamental interest in various areas of physical chemistry. In this contribution we have revisited the physisorption of hydrogen molecules and H2 clusters on fullerenes, following a first-principles approach in which the interaction is quantitatively evaluated for the C20 system using high-level electronic structure methods. Relative to coupled cluster data at the level of single, double, and perturbative triple excitations taken as a benchmark, the results for rotationally averaged physisorbed H2 show a good performance of MP2 variants and symmetry-adapted perturbation theory, but significant deviations and basis set convergence issues are found for dispersion-corrected density functional theory. These electronic structure data are fitted to produce effective coarse-grained potentials for use in larger systems such as C60–H2. Using path-integral molecular dynamics, the potentials are also applied to parahydrogen clusters solvated around fullerenes, across the regime where the first solvation shell becomes complete and as a function of increasing temperature. For C60 our findings indicate a sensible dependence of the critical solvation size on the underlying potential. As the temperature is increased, a competition is found between the surface and radial expansions of the solvation shell, with one molecule popping away at intermediate temperatures but getting reinserted at even higher temperatures.
中文翻译:

H 2在富勒烯上的物理吸附和氢簇在有限温度下对C 60的溶剂化:理论评估
氢和碳纳米结构之间的相互作用在物理化学的各个领域中具有根本的意义。在这一贡献中,我们遵循了第一原理方法,其中使用高级电子结构方法对C 20系统的相互作用进行了定量评估,从而重新研究了富勒烯上氢分子和H 2团簇的物理吸附。相对于以单,双和微扰三重激发水平为基础的耦合簇数据,以旋转平均的物理吸附H 2的结果为基准展示了MP2变体和对称自适应扰动理论的良好性能,但是色散校正的密度泛函理论发现了明显的偏差和基集收敛问题。对这些电子结构数据进行拟合,以产生有效的粗粒度电势,以用于较大的系统,例如C 60 -H 2。通过使用路径积分分子动力学,还可以将势能应用于富勒烯周围溶剂化的对氢簇,并跨越第一个溶剂化壳变得完整并随温度升高而变化的状态。对于C 60我们的发现表明临界溶剂化大小对潜在潜能的合理依赖。随着温度升高,在溶剂化壳的表面和径向膨胀之间发现竞争,一个分子在中间温度下突然消失,但在更高的温度下又重新插入。
更新日期:2018-02-16
中文翻译:
H 2在富勒烯上的物理吸附和氢簇在有限温度下对C 60的溶剂化:理论评估
氢和碳纳米结构之间的相互作用在物理化学的各个领域中具有根本的意义。在这一贡献中,我们遵循了第一原理方法,其中使用高级电子结构方法对C 20系统的相互作用进行了定量评估,从而重新研究了富勒烯上氢分子和H 2团簇的物理吸附。相对于以单,双和微扰三重激发水平为基础的耦合簇数据,以旋转平均的物理吸附H 2的结果为基准展示了MP2变体和对称自适应扰动理论的良好性能,但是色散校正的密度泛函理论发现了明显的偏差和基集收敛问题。对这些电子结构数据进行拟合,以产生有效的粗粒度电势,以用于较大的系统,例如C 60 -H 2。通过使用路径积分分子动力学,还可以将势能应用于富勒烯周围溶剂化的对氢簇,并跨越第一个溶剂化壳变得完整并随温度升高而变化的状态。对于C 60我们的发现表明临界溶剂化大小对潜在潜能的合理依赖。随着温度升高,在溶剂化壳的表面和径向膨胀之间发现竞争,一个分子在中间温度下突然消失,但在更高的温度下又重新插入。










































 京公网安备 11010802027423号
京公网安备 11010802027423号