当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Accurate Lattice Energies for Molecular Crystals from Experimental Crystal Structures
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2018-02-06 00:00:00 , DOI: 10.1021/acs.jctc.7b01200 Sajesh P. Thomas 1 , Peter R. Spackman 1 , Dylan Jayatilaka 1 , Mark A. Spackman 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2018-02-06 00:00:00 , DOI: 10.1021/acs.jctc.7b01200 Sajesh P. Thomas 1 , Peter R. Spackman 1 , Dylan Jayatilaka 1 , Mark A. Spackman 1
Affiliation
|
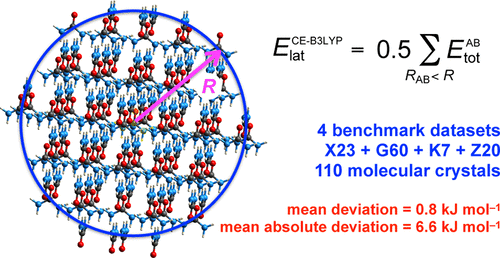
Using four different benchmark sets of molecular crystals, we establish the level of confidence for lattice energies estimated using CE-B3LYP model energies and experimental crystal structures. [ IUCrJ 2017, 4, 575−58710.1107/S205225251700848X.] We conclude that they compare very well with available benchmark estimates derived from sublimation enthalpies, and in many cases they are comparable with, and sometimes better than, more computationally demanding approaches, such as those based on periodic DFT plus dispersion methodologies. The performance over the complete set of 110 crystals indicates a mean absolute deviation from benchmark energies of only 6.6 kJ mol–1. Applications to polymorphic crystals and larger molecules are also presented and critically discussed. The results highlight the importance of recognizing the consequences of different sets of crystal/molecule geometries when different methodologies are compared, as well as the need for more extensive benchmark sets of crystal structures and associated lattice energies.
中文翻译:

从实验晶体结构对分子晶体的准确晶格能量
使用四个不同的基准分子晶体集,我们建立了使用CE-B3LYP模型能量和实验晶体结构估算的晶格能量的置信度。[ IUCrJ 2017年,4,575-58710.1107 / S205225251700848X]我们的结论是,他们比较非常好,从升华焓获得的可用基准估算,在许多情况下,它们相媲美,有时比,计算量大的方法,如基于定期DFT加上分散方法的方法。整套110晶体的性能表明,与基准能量的平均绝对偏差仅为6.6 kJ mol –1。还介绍并严格讨论了多晶型晶体和较大分子的应用。结果强调了比较不同方法时识别不同组晶体/分子几何结构的后果的重要性,以及对更广泛的晶体结构和相关晶格能基准组的需求。
更新日期:2018-02-06
中文翻译:
从实验晶体结构对分子晶体的准确晶格能量
使用四个不同的基准分子晶体集,我们建立了使用CE-B3LYP模型能量和实验晶体结构估算的晶格能量的置信度。[ IUCrJ 2017年,4,575-58710.1107 / S205225251700848X]我们的结论是,他们比较非常好,从升华焓获得的可用基准估算,在许多情况下,它们相媲美,有时比,计算量大的方法,如基于定期DFT加上分散方法的方法。整套110晶体的性能表明,与基准能量的平均绝对偏差仅为6.6 kJ mol –1。还介绍并严格讨论了多晶型晶体和较大分子的应用。结果强调了比较不同方法时识别不同组晶体/分子几何结构的后果的重要性,以及对更广泛的晶体结构和相关晶格能基准组的需求。








































 京公网安备 11010802027423号
京公网安备 11010802027423号