当前位置:
X-MOL 学术
›
J. Phys. Chem. B
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Molecular Dynamics Study of the Solution Structure, Clustering, and Diffusion of Four Aqueous Alkanolamines
The Journal of Physical Chemistry B ( IF 2.8 ) Pub Date : 2018-02-15 00:00:00 , DOI: 10.1021/acs.jpcb.7b10322 Sergey M. Melnikov 1 , Matthias Stein 1
The Journal of Physical Chemistry B ( IF 2.8 ) Pub Date : 2018-02-15 00:00:00 , DOI: 10.1021/acs.jpcb.7b10322 Sergey M. Melnikov 1 , Matthias Stein 1
Affiliation
|
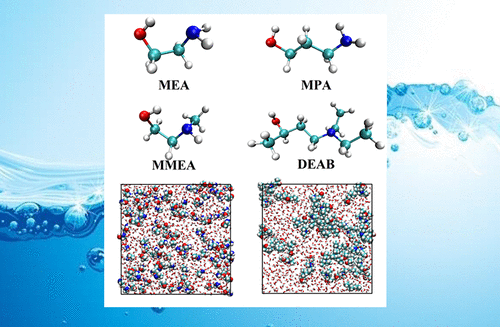
CO2 sequestration from anthropogenic resources is a challenge to the design of environmental processes at a large scale. Reversible chemical absorption by amine-based solvents is one of the most efficient methods of CO2 removal. Molecular simulation techniques are very useful tools to investigate CO2 binding by aqueous alkanolamine molecules for further technological application. In the present work, we have performed detailed atomistic molecular dynamics simulations of aqueous solutions of three prototype amines: monoethanolamine (MEA) as a standard, 3-aminopropanol (MPA), 2-methylaminoethanol (MMEA), and 4-diethylamino-2-butanol (DEAB) as potential novel CO2 absorptive solvents. Solvent densities, radial distribution functions, cluster size distributions, hydrogen-bonding statistics, and diffusion coefficients for a full range of mixture compositions have been obtained. The solvent densities and diffusion coefficients from simulations are in good agreement with those in the experiment. In aqueous solution, MEA, MPA, and MMEA molecules prefer to be fully solvated by water molecules, whereas DEAB molecules tend to self-aggregate. In a range from 30/70–50/50 (w/w) alkanolamine/water mixtures, they form a bicontinuous phase (both alkanolamine and water are organized in two mutually percolating clusters). Among the studied aqueous alkanolamine solutions, the diffusion coefficients decrease in the following order MEA > MPA = MMEA > DEAB. With an increase of water content, the diffusion coefficients increase for all studied alkanolamines. The presented results are a first step for process-scale simulation and provide important qualitative and quantitative information for the design and engineering of efficient new CO2 removal processes.
中文翻译:

四种链烷醇胺的溶液结构,聚集和扩散的分子动力学研究
从人为资源中隔离CO 2是大规模设计环境过程的挑战。胺基溶剂可逆的化学吸收是最有效的CO 2去除方法之一。分子模拟技术是研究含水链烷醇胺分子与CO 2结合以进行进一步技术应用的非常有用的工具。在目前的工作中,我们对三种原型胺的水溶液进行了详细的原子分子动力学模拟:单乙醇胺(MEA)作为标准溶液,3-氨基丙醇(MPA),2-甲基氨基乙醇(MMEA)和4-二乙基氨基-2-丁醇(DEAB)作为潜在的新型CO 2吸收性溶剂。已经获得了整个混合物组成范围的溶剂密度,径向分布函数,簇尺寸分布,氢键统计量和扩散系数。模拟得出的溶剂密度和扩散系数与实验中的一致。在水溶液中,MEA,MPA和MMEA分子倾向于被水分子完全溶解,而DEAB分子倾向于自聚集。链烷醇胺/水混合物的浓度范围为30 / 70–50 / 50(w / w),它们形成双连续相(链烷醇胺和水都组织在两个互相渗透的簇中)。在研究的链烷醇胺水溶液中,扩散系数按以下顺序递减:MEA> MPA = MMEA> DEAB。随着水含量的增加,所有研究的链烷醇胺的扩散系数均增加。给出的结果是过程规模仿真的第一步,并为高效的新CO的设计和工程提供重要的定性和定量信息。2个清除过程。
更新日期:2018-02-15
中文翻译:
四种链烷醇胺的溶液结构,聚集和扩散的分子动力学研究
从人为资源中隔离CO 2是大规模设计环境过程的挑战。胺基溶剂可逆的化学吸收是最有效的CO 2去除方法之一。分子模拟技术是研究含水链烷醇胺分子与CO 2结合以进行进一步技术应用的非常有用的工具。在目前的工作中,我们对三种原型胺的水溶液进行了详细的原子分子动力学模拟:单乙醇胺(MEA)作为标准溶液,3-氨基丙醇(MPA),2-甲基氨基乙醇(MMEA)和4-二乙基氨基-2-丁醇(DEAB)作为潜在的新型CO 2吸收性溶剂。已经获得了整个混合物组成范围的溶剂密度,径向分布函数,簇尺寸分布,氢键统计量和扩散系数。模拟得出的溶剂密度和扩散系数与实验中的一致。在水溶液中,MEA,MPA和MMEA分子倾向于被水分子完全溶解,而DEAB分子倾向于自聚集。链烷醇胺/水混合物的浓度范围为30 / 70–50 / 50(w / w),它们形成双连续相(链烷醇胺和水都组织在两个互相渗透的簇中)。在研究的链烷醇胺水溶液中,扩散系数按以下顺序递减:MEA> MPA = MMEA> DEAB。随着水含量的增加,所有研究的链烷醇胺的扩散系数均增加。给出的结果是过程规模仿真的第一步,并为高效的新CO的设计和工程提供重要的定性和定量信息。2个清除过程。










































 京公网安备 11010802027423号
京公网安备 11010802027423号