当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Comparative CHARMM and AMOEBA Simulations of Lanthanide Hydration Energetics and Experimental Aqueous-Solution Structures
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2018-02-13 00:00:00 , DOI: 10.1021/acs.jctc.7b01018 Baofu Qiao 1 , S. Skanthakumar 1 , L. Soderholm 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2018-02-13 00:00:00 , DOI: 10.1021/acs.jctc.7b01018 Baofu Qiao 1 , S. Skanthakumar 1 , L. Soderholm 1
Affiliation
|
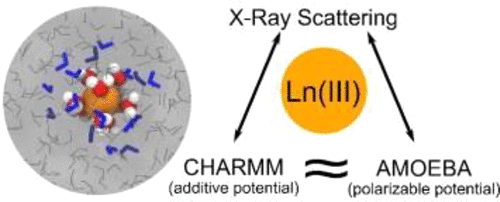
The accurate understanding of metal ion hydration in solutions is a prerequisite for predicting stability, reactivity, and solubility. Herein, additive CHARMM force field parameters were developed to enable molecular dynamics simulations of lanthanide (Ln) speciation in water. Quantitatively similar to the much more resource-intensive polarizable AMOEBA potential, the CHARMM simulations reproduce the experimental hydration free energies and correlations in the first shell (Ln-oxygen distance and hydration number). Comparisons of difference pair-distribution functions obtained from the two simulation approaches with those from high-energy X-ray scattering experiments reveal good agreement of first-coordination sphere correlations for the Lu3+ ion (CHARMM only), but further improvement to both approaches is required to reproduce the broad, non-Gaussian distribution seen from the La3+ experiment. Second-coordination sphere comparisons demonstrate the importance of explicitly including an anion in the simulation. This work describes the usefulness of less resource-intensive additive potentials in some complex chemical systems such as solution environments where multiple interactions have similar energetics. In addition, 3-dimensional descriptions of the La3+ and Lu3+ coordination geometries are extracted from the CHARMM simulations and generally discussed in terms of potential improvements to solute-structure modeling within solution environments.
中文翻译:

镧系元素水合能和实验水溶液结构的CHARMM和AMOEBA对比模拟
准确理解溶液中金属离子的水合作用是预测稳定性,反应性和溶解性的先决条件。在本文中,开发了附加的CHARMM力场参数,以实现水中镧系(Ln)物种形成的分子动力学模拟。CHARMM模拟在数量上与资源消耗更大的极化AMOEBA电位在数量上相似,在第一壳层中重现了实验的水合自由能及其相关性(Ln氧距离和水合数)。两种模拟方法获得的差分对分布函数与高能X射线散射实验获得的差分对分布函数的比较表明,Lu 3+的第一配位球相关性具有良好的一致性离子(仅适用于CHARMM),但需要进一步改进这两种方法,才能再现从La 3+实验中看到的宽泛的非高斯分布。第二配位球的比较证明了在模拟中明确包括阴离子的重要性。这项工作描述了在一些复杂的化学系统中,如多个相互作用具有相似能量的溶液环境中,资源消耗较少的添加剂势的有用性。此外,从CHARMM模拟中提取了La 3+和Lu 3+配位几何的3维描述,并通常根据对溶液环境中溶质结构建模的潜在改进进行讨论。
更新日期:2018-02-13
中文翻译:
镧系元素水合能和实验水溶液结构的CHARMM和AMOEBA对比模拟
准确理解溶液中金属离子的水合作用是预测稳定性,反应性和溶解性的先决条件。在本文中,开发了附加的CHARMM力场参数,以实现水中镧系(Ln)物种形成的分子动力学模拟。CHARMM模拟在数量上与资源消耗更大的极化AMOEBA电位在数量上相似,在第一壳层中重现了实验的水合自由能及其相关性(Ln氧距离和水合数)。两种模拟方法获得的差分对分布函数与高能X射线散射实验获得的差分对分布函数的比较表明,Lu 3+的第一配位球相关性具有良好的一致性离子(仅适用于CHARMM),但需要进一步改进这两种方法,才能再现从La 3+实验中看到的宽泛的非高斯分布。第二配位球的比较证明了在模拟中明确包括阴离子的重要性。这项工作描述了在一些复杂的化学系统中,如多个相互作用具有相似能量的溶液环境中,资源消耗较少的添加剂势的有用性。此外,从CHARMM模拟中提取了La 3+和Lu 3+配位几何的3维描述,并通常根据对溶液环境中溶质结构建模的潜在改进进行讨论。










































 京公网安备 11010802027423号
京公网安备 11010802027423号