当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Molecular Dynamics Simulations with Quantum Mechanics/Molecular Mechanics and Adaptive Neural Networks
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2018-02-13 00:00:00 , DOI: 10.1021/acs.jctc.7b01195 Lin Shen 1 , Weitao Yang 1, 2
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2018-02-13 00:00:00 , DOI: 10.1021/acs.jctc.7b01195 Lin Shen 1 , Weitao Yang 1, 2
Affiliation
|
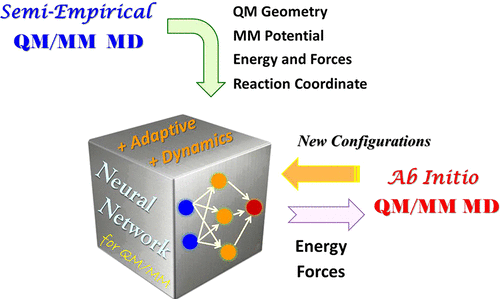
Direct molecular dynamics (MD) simulation with ab initio quantum mechanical and molecular mechanical (QM/MM) methods is very powerful for studying the mechanism of chemical reactions in a complex environment but also very time-consuming. The computational cost of QM/MM calculations during MD simulations can be reduced significantly using semiempirical QM/MM methods with lower accuracy. To achieve higher accuracy at the ab initio QM/MM level, a correction on the existing semiempirical QM/MM model is an attractive idea. Recently, we reported a neural network (NN) method as QM/MM-NN to predict the potential energy difference between semiempirical and ab initio QM/MM approaches. The high-level results can be obtained using neural network based on semiempirical QM/MM MD simulations, but the lack of direct MD samplings at the ab initio QM/MM level is still a deficiency that limits the applications of QM/MM-NN. In the present paper, we developed a dynamic scheme of QM/MM-NN for direct MD simulations on the NN-predicted potential energy surface to approximate ab initio QM/MM MD. Since some configurations excluded from the database for NN training were encountered during simulations, which may cause some difficulties on MD samplings, an adaptive procedure inspired by the selection scheme reported by Behler [Behler Int. J. Quantum Chem. 2015, 115, 1032; Behler Angew. Chem., Int. Ed. 2017, 56, 12828] was employed with some adaptions to update NN and carry out MD iteratively. We further applied the adaptive QM/MM-NN MD method to the free energy calculation and transition path optimization on chemical reactions in water. The results at the ab initio QM/MM level can be well reproduced using this method after 2–4 iteration cycles. The saving in computational cost is about 2 orders of magnitude. It demonstrates that the QM/MM-NN with direct MD simulations has great potentials not only for the calculation of thermodynamic properties but also for the characterization of reaction dynamics, which provides a useful tool to study chemical or biochemical systems in solution or enzymes.
中文翻译:

量子力学/分子力学和自适应神经网络的分子动力学模拟
使用从头算量子力学和分子力学 (QM/MM) 方法的直接分子动力学 (MD) 模拟对于研究复杂环境中的化学反应机制非常强大,但也非常耗时。使用精度较低的半经验 QM/MM 方法可以显着降低 MD 模拟期间 QM/MM 计算的计算成本。为了在从头开始的 QM/MM 水平上实现更高的精度,对现有的半经验 QM/MM 模型进行修正是一个有吸引力的想法。最近,我们报告了一种称为 QM/MM-NN 的神经网络 (NN) 方法,用于预测半经验方法和从头算 QM/MM 方法之间的势能差异。使用基于半经验 QM/MM MD 模拟的神经网络可以获得高水平的结果,但缺乏从头开始 QM/MM 级别的直接 MD 采样仍然是限制 QM/MM-NN 应用的缺陷。在本文中,我们开发了一种 QM/MM-NN 动态方案,用于在 NN 预测的势能表面上进行直接 MD 模拟,以近似从头开始的 QM/MM MD。由于在模拟过程中遇到了一些从数据库中排除的用于神经网络训练的配置,这可能会给 MD 采样带来一些困难,因此受 Behler [Behler Int. J.量子化学。 2015、115、1032;_ _ 贝勒·安吉奥. 化学,国际。埃德。 2017 , 56 , 12828] 进行了一些调整来更新 NN 并迭代执行 MD。我们进一步将自适应QM/MM-NN MD方法应用于水中化学反应的自由能计算和跃迁路径优化。在 2-4 个迭代周期后,使用此方法可以很好地重现从头开始 QM/MM 级别的结果。计算成本节省约 2 个数量级。它表明,直接MD模拟的QM/MM-NN不仅在热力学性质的计算方面具有巨大的潜力,而且在反应动力学的表征方面也具有巨大的潜力,这为研究溶液或酶中的化学或生化系统提供了有用的工具。
更新日期:2018-02-13
中文翻译:
量子力学/分子力学和自适应神经网络的分子动力学模拟
使用从头算量子力学和分子力学 (QM/MM) 方法的直接分子动力学 (MD) 模拟对于研究复杂环境中的化学反应机制非常强大,但也非常耗时。使用精度较低的半经验 QM/MM 方法可以显着降低 MD 模拟期间 QM/MM 计算的计算成本。为了在从头开始的 QM/MM 水平上实现更高的精度,对现有的半经验 QM/MM 模型进行修正是一个有吸引力的想法。最近,我们报告了一种称为 QM/MM-NN 的神经网络 (NN) 方法,用于预测半经验方法和从头算 QM/MM 方法之间的势能差异。使用基于半经验 QM/MM MD 模拟的神经网络可以获得高水平的结果,但缺乏从头开始 QM/MM 级别的直接 MD 采样仍然是限制 QM/MM-NN 应用的缺陷。在本文中,我们开发了一种 QM/MM-NN 动态方案,用于在 NN 预测的势能表面上进行直接 MD 模拟,以近似从头开始的 QM/MM MD。由于在模拟过程中遇到了一些从数据库中排除的用于神经网络训练的配置,这可能会给 MD 采样带来一些困难,因此受 Behler [Behler Int. J.量子化学。 2015、115、1032;_ _ 贝勒·安吉奥. 化学,国际。埃德。 2017 , 56 , 12828] 进行了一些调整来更新 NN 并迭代执行 MD。我们进一步将自适应QM/MM-NN MD方法应用于水中化学反应的自由能计算和跃迁路径优化。在 2-4 个迭代周期后,使用此方法可以很好地重现从头开始 QM/MM 级别的结果。计算成本节省约 2 个数量级。它表明,直接MD模拟的QM/MM-NN不仅在热力学性质的计算方面具有巨大的潜力,而且在反应动力学的表征方面也具有巨大的潜力,这为研究溶液或酶中的化学或生化系统提供了有用的工具。


























 京公网安备 11010802027423号
京公网安备 11010802027423号