当前位置:
X-MOL 学术
›
Ind. Eng. Chem. Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Thiophene Separation with Silver-Doped Cu-BTC Metal–Organic Framework for Deep Desulfurization
Industrial & Engineering Chemistry Research ( IF 3.8 ) Pub Date : 2018-02-19 00:00:00 , DOI: 10.1021/acs.iecr.7b04496 Shuai Ban 1 , Kaiyang Long 1 , Jing Xie 1 , Hui Sun 1 , Hongjun Zhou 1
Industrial & Engineering Chemistry Research ( IF 3.8 ) Pub Date : 2018-02-19 00:00:00 , DOI: 10.1021/acs.iecr.7b04496 Shuai Ban 1 , Kaiyang Long 1 , Jing Xie 1 , Hui Sun 1 , Hongjun Zhou 1
Affiliation
|
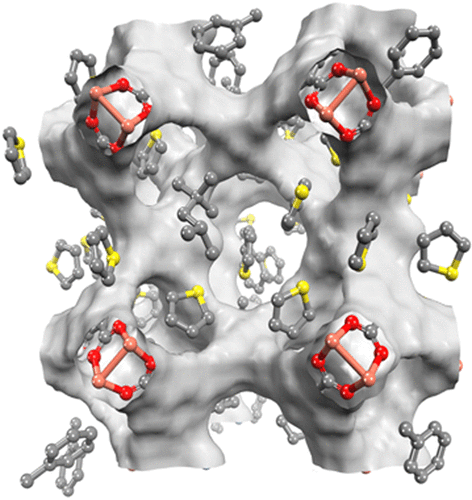
Molecular simulations using grand-canonical ensemble Monte Carlo (GCMC) and first-principles calculations were performed to study the silver doped copper(II) benzene-1,3,5-tricarboxylate (Cu-BTC) metal organic framework (MOF) for deep desulfurization. The density function theory (DFT) calculations were conducted to quantify the binding energies of silver dopant with Cu-BTC as well as adsorbates. The computed DFT potentials were taken to parametrize the classical host–guest force fields for GCMC simulations with high accuracy. Adsorption isotherms of thiophene and toluene in isooctane solutions were experimentally measured using pure Cu-BTC samples for the validation of molecular modeling. The simulation results show that the doping of silver leads to significant increase of adsorption uptakes for all components (thiophene, toluene, and isooctane), agreeing with the results of calculated adsorption enthalpies in Ag-Cu-BTC. Simulation snapshots reveal that thiophene is preferentially accommodated in the small tetrahedral cage of Cu-BTC, before intruding into the large octahedral cages to suppress loadings of toluene and isooctane with the assistance of silver dopants. The resulting selectivity of thiophene to toluene in isooctane was as high as 4 in Ag-Cu-BTC. On the basis of the mechanistic understandings, screen criteria and fictionalization schemes of potential MOF materials are proposed for applications of adsorptive desulfurization.
中文翻译:

掺杂银的Cu-BTC金属-有机骨架用于深脱硫的噻吩分离
使用大正则合奏蒙特卡洛(GCMC)分子模拟和第一性原理计算,研究了掺银的1,2,3,5-三羧酸铜(II)苯(Cu-BTC)金属有机骨架(MOF)的深度脱硫。进行了密度泛函理论(DFT)计算,以量化银掺杂剂与Cu-BTC以及被吸附物的结合能。计算出的DFT电位用于对经典的主客体力场进行参数化,以进行高精度的GCMC模拟。使用纯Cu-BTC样品通过实验测量了异辛烷溶液中噻吩和甲苯的吸附等温线,以验证分子模型。模拟结果表明,银的掺杂导致所有组分(噻吩,甲苯和异辛烷)的吸附吸收显着增加,与Ag-Cu-BTC中吸附焓的计算结果相符。模拟快照显示,噻吩优先容纳在Cu-BTC的小四面体笼中,然后侵入大的八面体笼中,以借助银掺杂剂抑制甲苯和异辛烷的负载。在Ag-Cu-BTC中,噻吩在异辛烷中对甲苯的选择性高达4。在对机理的理解的基础上,提出了潜在的MOF材料的筛选标准和虚拟化方案,以用于吸附脱硫的应用。在侵入大型八面体笼子之前,借助银掺杂剂抑制甲苯和异辛烷的负载。在Ag-Cu-BTC中,噻吩在异辛烷中对甲苯的选择性高达4。在对机理的理解的基础上,提出了潜在的MOF材料的筛选标准和虚拟化方案,以用于吸附脱硫的应用。在侵入大型八面体笼子之前,借助银掺杂剂抑制甲苯和异辛烷的负载。在Ag-Cu-BTC中,噻吩在异辛烷中对甲苯的选择性高达4。在对机理的理解的基础上,提出了潜在的MOF材料的筛选标准和虚拟化方案,以用于吸附脱硫的应用。
更新日期:2018-02-21
中文翻译:
掺杂银的Cu-BTC金属-有机骨架用于深脱硫的噻吩分离
使用大正则合奏蒙特卡洛(GCMC)分子模拟和第一性原理计算,研究了掺银的1,2,3,5-三羧酸铜(II)苯(Cu-BTC)金属有机骨架(MOF)的深度脱硫。进行了密度泛函理论(DFT)计算,以量化银掺杂剂与Cu-BTC以及被吸附物的结合能。计算出的DFT电位用于对经典的主客体力场进行参数化,以进行高精度的GCMC模拟。使用纯Cu-BTC样品通过实验测量了异辛烷溶液中噻吩和甲苯的吸附等温线,以验证分子模型。模拟结果表明,银的掺杂导致所有组分(噻吩,甲苯和异辛烷)的吸附吸收显着增加,与Ag-Cu-BTC中吸附焓的计算结果相符。模拟快照显示,噻吩优先容纳在Cu-BTC的小四面体笼中,然后侵入大的八面体笼中,以借助银掺杂剂抑制甲苯和异辛烷的负载。在Ag-Cu-BTC中,噻吩在异辛烷中对甲苯的选择性高达4。在对机理的理解的基础上,提出了潜在的MOF材料的筛选标准和虚拟化方案,以用于吸附脱硫的应用。在侵入大型八面体笼子之前,借助银掺杂剂抑制甲苯和异辛烷的负载。在Ag-Cu-BTC中,噻吩在异辛烷中对甲苯的选择性高达4。在对机理的理解的基础上,提出了潜在的MOF材料的筛选标准和虚拟化方案,以用于吸附脱硫的应用。在侵入大型八面体笼子之前,借助银掺杂剂抑制甲苯和异辛烷的负载。在Ag-Cu-BTC中,噻吩在异辛烷中对甲苯的选择性高达4。在对机理的理解的基础上,提出了潜在的MOF材料的筛选标准和虚拟化方案,以用于吸附脱硫的应用。










































 京公网安备 11010802027423号
京公网安备 11010802027423号