当前位置:
X-MOL 学术
›
J. Phys. Chem. B
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Pushing the Limits of Structure-Based Models: Prediction of Nonglobular Protein Folding and Fibrils Formation with Go-Model Simulations
The Journal of Physical Chemistry B ( IF 3.3 ) Pub Date : 2018-02-09 00:00:00 , DOI: 10.1021/acs.jpcb.7b12129 Lokesh Baweja 1 , Julien Roche 1
The Journal of Physical Chemistry B ( IF 3.3 ) Pub Date : 2018-02-09 00:00:00 , DOI: 10.1021/acs.jpcb.7b12129 Lokesh Baweja 1 , Julien Roche 1
Affiliation
|
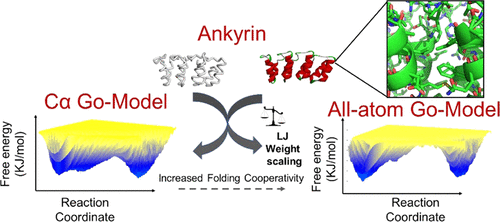
The development of computational efficient models is essential to obtain a detailed characterization of the mechanisms underlying the folding of proteins and the formation of amyloid fibrils. Structure-based computational models (Go-model) with Cα or all-atom resolutions have been able to successfully delineate the mechanisms of folding of several globular proteins and offer an interesting alternative to computationally intensive simulations with explicit solvent description. Here, we explore the limits of Go-model predictions by analyzing the folding of the nonglobular repeat domain proteins Notch Ankyrin and p16INK4 and the formation of human islet amyloid polypeptide (hIAPP) fibrils. Folding trajectories of the repeat domain proteins revealed that an all-atom resolution is required to capture the folding pathways and cooperativity reported in experimental studies. The all-atom Go-model was also successful in predicting the free-energy landscape of hIAPP fibrillation, suggesting a “dock and lock” mechanism of fibril elongation. We finally explored how mutations can affect the co-assembly of hIAPP fibrils by simulating a heterogeneous system composed of wild-type and mutated hIAPP peptides. Overall, this study shows that all-atom Go-model-based simulations have the potential of discerning the effects of mutations and post-translational modifications in protein folding and association and may help in resolving the dichotomy between experimental and theoretical studies on protein folding and amyloid fibrillation.
中文翻译:

突破基于结构的模型的局限性:Go模型模拟预测小叶蛋白折叠和原纤维形成
计算有效模型的开发对于获得蛋白质折叠和淀粉样蛋白原纤维形成机理的详细表征至关重要。具有Cα或全原子分辨率的基于结构的计算模型(Go模型)已经能够成功地描绘出几种球状蛋白质折叠的机制,并为具有明确溶剂描述的计算密集型模拟提供了有趣的替代方法。在这里,我们通过分析非球形重复域蛋白Notch Ankyrin和p16 INK4的折叠来探索Go模型预测的局限性并形成人胰岛淀粉样多肽(hIAPP)原纤维。重复域蛋白的折叠轨迹表明,需要全原子分辨率才能捕获实验研究中报道的折叠途径和协同作用。全原子Go模型还成功地预测了hIAPP纤颤的自由能态势,表明了纤丝伸长的“对接和锁定”机制。最后,我们通过模拟由野生型和突变的hIAPP肽组成的异质系统,探讨了突变如何影响hIAPP原纤维的共装配。全面的,
更新日期:2018-02-09
中文翻译:
突破基于结构的模型的局限性:Go模型模拟预测小叶蛋白折叠和原纤维形成
计算有效模型的开发对于获得蛋白质折叠和淀粉样蛋白原纤维形成机理的详细表征至关重要。具有Cα或全原子分辨率的基于结构的计算模型(Go模型)已经能够成功地描绘出几种球状蛋白质折叠的机制,并为具有明确溶剂描述的计算密集型模拟提供了有趣的替代方法。在这里,我们通过分析非球形重复域蛋白Notch Ankyrin和p16 INK4的折叠来探索Go模型预测的局限性并形成人胰岛淀粉样多肽(hIAPP)原纤维。重复域蛋白的折叠轨迹表明,需要全原子分辨率才能捕获实验研究中报道的折叠途径和协同作用。全原子Go模型还成功地预测了hIAPP纤颤的自由能态势,表明了纤丝伸长的“对接和锁定”机制。最后,我们通过模拟由野生型和突变的hIAPP肽组成的异质系统,探讨了突变如何影响hIAPP原纤维的共装配。全面的,


























 京公网安备 11010802027423号
京公网安备 11010802027423号