当前位置:
X-MOL 学术
›
Energy Environ. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Band engineering of multicomponent semiconductors: a general theoretical model on the anion group†
Energy & Environmental Science ( IF 32.4 ) Pub Date : 2018-02-06 00:00:00 , DOI: 10.1039/c7ee03503a X. Y. Meng 1, 2, 3, 4, 5 , D. Y. Liu 1, 2, 3, 4 , G. W. Qin 2, 3, 4, 5, 6
Energy & Environmental Science ( IF 32.4 ) Pub Date : 2018-02-06 00:00:00 , DOI: 10.1039/c7ee03503a X. Y. Meng 1, 2, 3, 4, 5 , D. Y. Liu 1, 2, 3, 4 , G. W. Qin 2, 3, 4, 5, 6
Affiliation
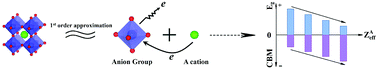
|
Development of energy conversion semiconductor materials has attracted increasing interest over the past three decades, but most successful semiconductors are unary or binary, rather than multicomponent semiconductors (MCSCs). There is a several orders of magnitude wider variety of MCSCs than unary and binary semiconductors, but very few electronic energy theories have been able to deal with more than two composition variables so far, and thus desired MCSCs are hard to predict. In this work, we propose a universal anion group model based on the analysis of electronic structures in an ABO3 perovskite prototype. Under a first order approximation, that is, the ‘A’ cation and the (BO6) anion group have very little hybridization, we find that the band gap of the ABO3 semiconductor is mainly determined by the (BO6) anion group and is very similar to that of binary compounds consisting of the same anion group constituents, while the band edges can be adjusted by the ‘A’-site cation. When more intense hybridizations exist, the predicted results can be amended by considering the higher order approximation. Using this model, the band gaps and edges of quaternary AgxNa(1−x)NbO3 perovskites and ZnxMg(1−x)Fe2O4 spinels have been predicted and are consistent with reported experiments and first principles calculations, further confirming the validity of the proposed model. Therefore, an anion group model on MCSCs can not only promote the probability of success in band engineering, but can also pave the way for speeding up the design of novel and desired MCSCs using known binary semiconductors for use in the field of energy conversion materials as photocatalysts, light-emitting materials, complementary light-absorption materials for solar cells, etc.
中文翻译:

多组分半导体的能带工程:有关阴离子基团的一般理论模型†
在过去的三十年中,能量转换半导体材料的开发引起了越来越多的兴趣,但是大多数成功的半导体都是一元或二元的,而不是多组分半导体(MCSC)。与一元和二元半导体相比,MCSC的变化范围大几个数量级,但是到目前为止,很少有电子能量理论能够处理两个以上的组成变量,因此很难预测所需的MCSC。在这项工作中,我们提出了一个基于ABO 3钙钛矿原型中电子结构分析的通用阴离子基团模型。在一级近似下,即'A'阳离子和(BO 6)阴离子基团的杂交非常少,我们发现ABO 3的带隙半导体主要由(BO 6)阴离子基团决定,并且与由相同阴离子基团组成的二元化合物非常相似,而能带边缘可通过'A'位阳离子进行调节。当存在更强烈的杂交时,可以通过考虑更高阶的逼近来修改预测结果。使用该模型,季铵Ag x Na (1- x) NbO 3钙钛矿和Zn x Mg (1- x) Fe 2 O 4的带隙和边缘尖晶石已经被预测并与报道的实验和第一性原理计算相一致,进一步证实了所提出模型的有效性。因此,基于MCSC的阴离子基团模型不仅可以提高谱带工程成功的可能性,而且还可以为在能量转换材料领域中使用已知的二元半导体加快新型和期望的MCSC的设计铺平道路。光催化剂,发光材料,太阳能电池的辅助吸光材料等。
更新日期:2018-02-06
中文翻译:
多组分半导体的能带工程:有关阴离子基团的一般理论模型†
在过去的三十年中,能量转换半导体材料的开发引起了越来越多的兴趣,但是大多数成功的半导体都是一元或二元的,而不是多组分半导体(MCSC)。与一元和二元半导体相比,MCSC的变化范围大几个数量级,但是到目前为止,很少有电子能量理论能够处理两个以上的组成变量,因此很难预测所需的MCSC。在这项工作中,我们提出了一个基于ABO 3钙钛矿原型中电子结构分析的通用阴离子基团模型。在一级近似下,即'A'阳离子和(BO 6)阴离子基团的杂交非常少,我们发现ABO 3的带隙半导体主要由(BO 6)阴离子基团决定,并且与由相同阴离子基团组成的二元化合物非常相似,而能带边缘可通过'A'位阳离子进行调节。当存在更强烈的杂交时,可以通过考虑更高阶的逼近来修改预测结果。使用该模型,季铵Ag x Na (1- x) NbO 3钙钛矿和Zn x Mg (1- x) Fe 2 O 4的带隙和边缘尖晶石已经被预测并与报道的实验和第一性原理计算相一致,进一步证实了所提出模型的有效性。因此,基于MCSC的阴离子基团模型不仅可以提高谱带工程成功的可能性,而且还可以为在能量转换材料领域中使用已知的二元半导体加快新型和期望的MCSC的设计铺平道路。光催化剂,发光材料,太阳能电池的辅助吸光材料等。










































 京公网安备 11010802027423号
京公网安备 11010802027423号