当前位置:
X-MOL 学术
›
J. Proteome Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
A Comparison of Common Mass Spectrometry Approaches for Paleoproteomics
Journal of Proteome Research ( IF 4.4 ) Pub Date : 2018-01-31 00:00:00 , DOI: 10.1021/acs.jproteome.7b00703 Timothy P. Cleland 1 , Elena R. Schroeter 2
Journal of Proteome Research ( IF 4.4 ) Pub Date : 2018-01-31 00:00:00 , DOI: 10.1021/acs.jproteome.7b00703 Timothy P. Cleland 1 , Elena R. Schroeter 2
Affiliation
|
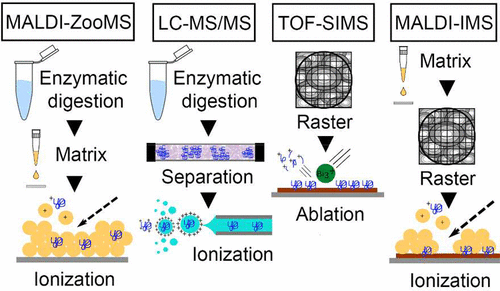
The last two decades have seen a broad diversity of methods used to identify and/or characterize proteins in the archeological and paleontological record. Of these, mass spectrometry has opened an unprecedented window into the proteomes of the past, providing protein sequence data from long extinct animals as well as historical and prehistorical artifacts. Thus, application of mass spectrometry to fossil remains has become an attractive source for ancient molecular sequences with which to conduct evolutionary studies, particularly in specimens older than the proposed limit of amplifiable DNA detection. However, “mass spectrometry” covers a range of mass-based proteomic approaches, each of which utilize different technology and physical principles to generate unique types of data, with their own strengths and challenges. Here, we discuss a variety of mass spectrometry techniques that have or may be used to detect and characterize archeological and paleontological proteins, with a particular focus on MALDI-MS, LC–MS/MS, TOF-SIMS, and MSi. The main differences in their functionality, the types of data they produce, and the potential effects of diagenesis on their results are considered.
中文翻译:

普通蛋白质组学质谱方法的比较
在过去的二十年中,已经发现了用于鉴定和/或表征考古和古生物学记录中蛋白质的方法的多样性。其中,质谱仪为过去的蛋白质组学打开了前所未有的窗口,提供了来自长期灭绝的动物以及历史和史前人工制品的蛋白质序列数据。因此,将质谱法应用于化石遗迹已成为进行进化研究的古老分子序列的一种有吸引力的来源,尤其是在比拟议的可扩增DNA检测极限更老的标本中。但是,“质谱法”涵盖了一系列基于质量的蛋白质组学方法,每种方法都利用不同的技术和物理原理来生成独特类型的数据,并具有自己的优势和挑战。这里,我们讨论了已经或可能用于检测和表征考古和古生物学蛋白质的各种质谱技术,尤其侧重于MALDI-MS,LC-MS / MS,TOF-SIMS和MSi。考虑了它们的功能,产生的数据类型以及成岩作用对其结果的潜在影响方面的主要差异。
更新日期:2018-01-31
中文翻译:
普通蛋白质组学质谱方法的比较
在过去的二十年中,已经发现了用于鉴定和/或表征考古和古生物学记录中蛋白质的方法的多样性。其中,质谱仪为过去的蛋白质组学打开了前所未有的窗口,提供了来自长期灭绝的动物以及历史和史前人工制品的蛋白质序列数据。因此,将质谱法应用于化石遗迹已成为进行进化研究的古老分子序列的一种有吸引力的来源,尤其是在比拟议的可扩增DNA检测极限更老的标本中。但是,“质谱法”涵盖了一系列基于质量的蛋白质组学方法,每种方法都利用不同的技术和物理原理来生成独特类型的数据,并具有自己的优势和挑战。这里,我们讨论了已经或可能用于检测和表征考古和古生物学蛋白质的各种质谱技术,尤其侧重于MALDI-MS,LC-MS / MS,TOF-SIMS和MSi。考虑了它们的功能,产生的数据类型以及成岩作用对其结果的潜在影响方面的主要差异。


























 京公网安备 11010802027423号
京公网安备 11010802027423号