当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Fully Flexible Docking via Reaction-Coordinate-Independent Molecular Dynamics Simulations
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2018-02-09 00:00:00 , DOI: 10.1021/acs.jcim.7b00674 Martina Bertazzo 1, 2 , Mattia Bernetti 1, 2 , Maurizio Recanatini 1 , Matteo Masetti 1 , Andrea Cavalli 1, 2
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2018-02-09 00:00:00 , DOI: 10.1021/acs.jcim.7b00674 Martina Bertazzo 1, 2 , Mattia Bernetti 1, 2 , Maurizio Recanatini 1 , Matteo Masetti 1 , Andrea Cavalli 1, 2
Affiliation
|
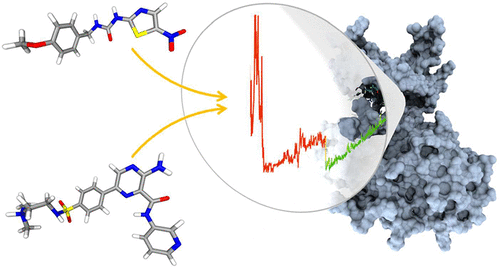
Predicting the geometry of protein–ligand binding complexes is of primary importance for structure-based drug discovery. Molecular dynamics (MD) is emerging as a reliable computational tool for use in conjunction with, or an alternative to, docking methods. However, simulating the protein–ligand binding process often requires very expensive simulations. This drastically limits the practical application of MD-based approaches. Here, we propose a general framework to accelerate the generation of putative protein–ligand binding modes using potential-scaled MD simulations. The proposed dynamical protocol has been applied to two pharmaceutically relevant systems (GSK-3β and the N-terminal domain of HSP90α). Our approach is fully independent of any predefined reaction coordinate (or collective variable). It identified the correct binding mode of several ligands and can thus save valuable computational time in dynamic docking simulations.
中文翻译:

通过与反应坐标无关的分子动力学模拟进行完全灵活的对接
预测蛋白质-配体结合复合物的几何形状对于基于结构的药物发现至关重要。分子动力学(MD)逐渐成为一种可靠的计算工具,可与对接方法结合使用或替代对接方法。但是,模拟蛋白质-配体结合过程通常需要非常昂贵的模拟。这极大地限制了基于MD的方法的实际应用。在这里,我们提出了使用潜在规模的MD模拟来加速推定的蛋白质-配体结合模式生成的通用框架。拟议的动态协议已应用于两个药物相关的系统(GSK-3β和HSP90α的N末端域)。我们的方法完全独立于任何预定义的反应坐标(或集合变量)。
更新日期:2018-02-09
中文翻译:
通过与反应坐标无关的分子动力学模拟进行完全灵活的对接
预测蛋白质-配体结合复合物的几何形状对于基于结构的药物发现至关重要。分子动力学(MD)逐渐成为一种可靠的计算工具,可与对接方法结合使用或替代对接方法。但是,模拟蛋白质-配体结合过程通常需要非常昂贵的模拟。这极大地限制了基于MD的方法的实际应用。在这里,我们提出了使用潜在规模的MD模拟来加速推定的蛋白质-配体结合模式生成的通用框架。拟议的动态协议已应用于两个药物相关的系统(GSK-3β和HSP90α的N末端域)。我们的方法完全独立于任何预定义的反应坐标(或集合变量)。










































 京公网安备 11010802027423号
京公网安备 11010802027423号