Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Mapping Causal Variants with Single-Nucleotide Resolution Reveals Biochemical Drivers of Phenotypic Change.
Cell ( IF 64.5 ) Pub Date : 2018-Jan-25 , DOI: 10.1016/j.cell.2017.12.015 Richard She 1 , Daniel F Jarosz 2
Cell ( IF 64.5 ) Pub Date : 2018-Jan-25 , DOI: 10.1016/j.cell.2017.12.015 Richard She 1 , Daniel F Jarosz 2
Affiliation
|
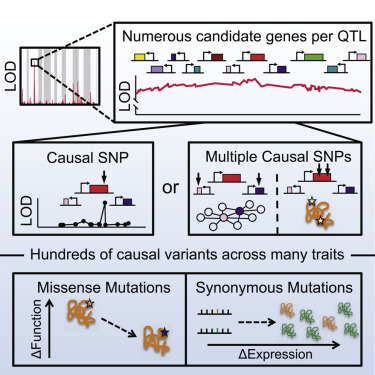
Understanding the sequence determinants that give rise to diversity among individuals and species is the central challenge of genetics. However, despite ever greater numbers of sequenced genomes, most genome-wide association studies cannot distinguish causal variants from linked passenger mutations spanning many genes. We report that this inherent challenge can be overcome in model organisms. By pushing the advantages of inbred crossing to its practical limit in Saccharomyces cerevisiae, we improved the statistical resolution of linkage analysis to single nucleotides. This "super-resolution" approach allowed us to map 370 causal variants across 26 quantitative traits. Missense, synonymous, and cis-regulatory mutations collectively gave rise to phenotypic diversity, providing mechanistic insight into the basis of evolutionary divergence. Our data also systematically unmasked complex genetic architectures, revealing that multiple closely linked driver mutations frequently act on the same quantitative trait. Single-nucleotide mapping thus complements traditional deletion and overexpression screening paradigms and opens new frontiers in quantitative genetics.
中文翻译:

通过单核苷酸分辨率绘制因果变异图谱揭示了表型变化的生化驱动因素。
了解导致个体和物种多样性的序列决定因素是遗传学的核心挑战。然而,尽管测序的基因组数量越来越多,但大多数全基因组关联研究无法区分因果变异和跨越许多基因的连锁乘客突变。我们报告说,这种固有的挑战可以在模式生物中克服。通过将近交杂交的优势在酿酒酵母中发挥到实际极限,我们提高了单核苷酸连锁分析的统计分辨率。这种“超分辨率”方法使我们能够绘制 26 个数量性状的 370 个因果变异图。错义、同义和顺式调控突变共同产生了表型多样性,为进化分歧的基础提供了机制性的见解。我们的数据还系统地揭示了复杂的遗传结构,揭示了多个密切相关的驱动突变经常作用于相同的数量性状。因此,单核苷酸作图补充了传统的缺失和过度表达筛选范例,并开辟了定量遗传学的新领域。
更新日期:2018-01-25
中文翻译:
通过单核苷酸分辨率绘制因果变异图谱揭示了表型变化的生化驱动因素。
了解导致个体和物种多样性的序列决定因素是遗传学的核心挑战。然而,尽管测序的基因组数量越来越多,但大多数全基因组关联研究无法区分因果变异和跨越许多基因的连锁乘客突变。我们报告说,这种固有的挑战可以在模式生物中克服。通过将近交杂交的优势在酿酒酵母中发挥到实际极限,我们提高了单核苷酸连锁分析的统计分辨率。这种“超分辨率”方法使我们能够绘制 26 个数量性状的 370 个因果变异图。错义、同义和顺式调控突变共同产生了表型多样性,为进化分歧的基础提供了机制性的见解。我们的数据还系统地揭示了复杂的遗传结构,揭示了多个密切相关的驱动突变经常作用于相同的数量性状。因此,单核苷酸作图补充了传统的缺失和过度表达筛选范例,并开辟了定量遗传学的新领域。


























 京公网安备 11010802027423号
京公网安备 11010802027423号