当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Structural Articulation of Biochemical Reactions Using Restrained Geometries and Topology Switching
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2018-02-05 00:00:00 , DOI: 10.1021/acs.jcim.7b00699 Swati R. Manjari 1 , Nilesh K. Banavali 1, 2
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2018-02-05 00:00:00 , DOI: 10.1021/acs.jcim.7b00699 Swati R. Manjari 1 , Nilesh K. Banavali 1, 2
Affiliation
|
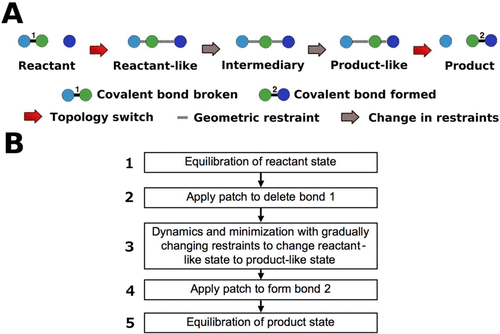
A strategy named “restrained geometries and topology switching” (RGATS) is presented to obtain detailed trajectories for complex biochemical reactions using molecular mechanics (MM) methods. It enables prediction of realistic dynamical pathways for chemical reactions, especially for accurately characterizing the structural adjustments of highly complex environments to any proximal biochemical reaction. It can be used to generate reactive conformations, model stepwise or concerted reactions in complex environments, and probe the influence of changes in the environment. Its ability to take reactively nonoptimal conformations and generate favorable starting conformations for a biochemical reaction is illustrated for a proton transfer between two model compounds. Its ability to study concerted reactions in explicit solvent is illustrated using proton transfers between an ammonium ion and two conserved histidines in an ammonia transporter channel embedded in a lipid membrane. Its ability to characterize the changes induced by subtle differences in the active site environment is illustrated using nucleotide addition by a DNA polymerase in the presence of two versus three Mg2+ ions. RGATS can be employed within any MM program and requires no additional software implementation. This allows the full assortment of computational methods implemented in all available MM programs to be used to tackle virtually any question about biochemical reactions that is answerable without using a quantum mechanical (QM) model. It can also be applied to generate reasonable starting structures for more detailed and expensive QM or QM/MM methods. In particular, this strategy enables rapid prediction of reactant, intermediary, or product state structures in any macromolecular context, with the only requirement being that the structure in any one of these states is either known or can be accurately modeled.
中文翻译:

使用约束的几何和拓扑转换的生化反应的结构表达。
提出了一种名为“约束几何和拓扑转换”(RGATS)的策略,以使用分子力学(MM)方法获得复杂生化反应的详细轨迹。它可以预测化学反应的现实动力学路径,尤其是对于准确表征高度复杂的环境对任何近端生化反应的结构调节而言。它可用于生成反应性构象,在复杂环境中逐步建模或协调一致的反应,并探究环境变化的影响。对于两种模型化合物之间的质子转移,说明了其采取反应性非最佳构象并产生有利于生化反应的起始构象的能力。通过在铵离子和嵌入脂质膜中的氨转运通道中的两个保守组氨酸之间进行质子转移,说明了其在显式溶剂中研究协同反应的能力。通过在2 Mg和3 Mg存在下通过DNA聚合酶添加核苷酸来说明其表征活性位点环境中细微差异引起的变化的能力2+离子。RGATS可以在任何MM程序中使用,并且不需要其他软件实现。这允许在所有可用的MM程序中实施的各种计算方法,几乎可以用来解决有关生化反应的任何问题,而无需使用量子力学(QM)模型即可解决。它也可以用于生成合理的起始结构,以用于更详细,更昂贵的QM或QM / MM方法。特别地,该策略能够在任何大分子环境中快速预测反应物,中间物或产物的状态结构,唯一的要求是这些状态中的任何一种的结构是已知的或可以被精确地建模。
更新日期:2018-02-05
中文翻译:
使用约束的几何和拓扑转换的生化反应的结构表达。
提出了一种名为“约束几何和拓扑转换”(RGATS)的策略,以使用分子力学(MM)方法获得复杂生化反应的详细轨迹。它可以预测化学反应的现实动力学路径,尤其是对于准确表征高度复杂的环境对任何近端生化反应的结构调节而言。它可用于生成反应性构象,在复杂环境中逐步建模或协调一致的反应,并探究环境变化的影响。对于两种模型化合物之间的质子转移,说明了其采取反应性非最佳构象并产生有利于生化反应的起始构象的能力。通过在铵离子和嵌入脂质膜中的氨转运通道中的两个保守组氨酸之间进行质子转移,说明了其在显式溶剂中研究协同反应的能力。通过在2 Mg和3 Mg存在下通过DNA聚合酶添加核苷酸来说明其表征活性位点环境中细微差异引起的变化的能力2+离子。RGATS可以在任何MM程序中使用,并且不需要其他软件实现。这允许在所有可用的MM程序中实施的各种计算方法,几乎可以用来解决有关生化反应的任何问题,而无需使用量子力学(QM)模型即可解决。它也可以用于生成合理的起始结构,以用于更详细,更昂贵的QM或QM / MM方法。特别地,该策略能够在任何大分子环境中快速预测反应物,中间物或产物的状态结构,唯一的要求是这些状态中的任何一种的结构是已知的或可以被精确地建模。


























 京公网安备 11010802027423号
京公网安备 11010802027423号