当前位置:
X-MOL 学术
›
ChemistrySelect
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Strategic Design and Utilization of Molecular Flexibility for Straddling the Application of Organic Superbases: A DFT Study
ChemistrySelect ( IF 1.9 ) Pub Date : 2018-01-16 , DOI: 10.1002/slct.201702912 Ajeet Singh 1, 2 , Animesh K. Ojha 1 , Hyun Myung Jang 2
ChemistrySelect ( IF 1.9 ) Pub Date : 2018-01-16 , DOI: 10.1002/slct.201702912 Ajeet Singh 1, 2 , Animesh K. Ojha 1 , Hyun Myung Jang 2
Affiliation
|
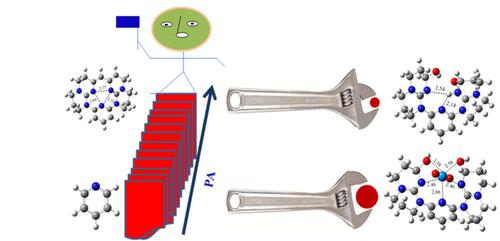
The density functional theory (DFT) calculations were performed for a series of new molecular frameworks that have potential to work as organic superbases. In the present report, we have exploited and strategically substituted 2 and 6 positions of the pyridine by a potential anchoring group, 1,5,7‐triazabicyclo[4.4.0]dec‐5‐ene (TBD). The value of proton affinities (PAs) of the molecular frameworks was calculated in the gas phase as well as in acetonitrile solution. The designed non‐ionic and neutral organic superbases were found to have higher basicity than that of the benchmarked molecule, 1,8‐bis(dimethylamino)‐naphthalene (DMAN). The zero‐point vibrational energy (ZPVE) and PAs values were calculated for the designed molecular frameworks at B3LYP/6‐311+G**//B3LYP/6‐31+G* level of theory. For compression, we have also performed the optimization of all the structures at M06‐2X/6‐311 +G**//M06‐2X/6‐31+G* level of theory. The molecular framework 9 has highest PAs values 1151.2 and 1246.3 kJ mol−1 in the gas phase and in acetonitrile solution, respectively at B3LYP/6‐311+G**//B3LYP/6‐31+G* level of theory. The designed molecular frameworks have better flexibility, which enables it for selective extraction of the smaller to larger size molecules by varying the size of the cavity, as required for the various applications. In this context, we have explored the application of designed molecular frameworks for the selective extraction of UO22+ over VO2+.
中文翻译:

跨越有机超强碱应用的分子柔性的策略设计和利用:DFT研究
密度泛函理论(DFT)计算是针对一系列可能用作有机超碱的新型分子框架进行的。在本报告中,我们利用潜在的锚定基团1,5,7-三氮杂双环[4.4.0] dec-5-ene(TBD)开发并战略性地取代了吡啶的2和6位。计算了气相以及乙腈溶液中分子骨架的质子亲和力(PAs)值。发现设计的非离子和中性有机超碱具有比基准分子1,8-双(二甲基氨基)-萘(DMAN)更高的碱度。在理论水平为B3LYP / 6-311 + G ** // B3LYP / 6-31 + G *的情况下,针对设计的分子框架计算了零点振动能(ZPVE)和PAs值。对于压缩,我们还在M06-2X / 6-311 + G ** // M06-2X / 6-31 + G *理论水平上对所有结构进行了优化。分子框架图9在气相和乙腈溶液中的理论PA值分别最高为1151.2和1246.3 kJ mol -1,分别为B3LYP / 6-311 + G ** // B3LYP / 6-31 + G *。设计的分子框架具有更好的柔韧性,使其能够根据各种应用的需要,通过改变腔体的大小来选择性地提取较小至较大尺寸的分子。在这种情况下,我们探索了设计的分子框架在VO 2 +上选择性萃取UO 2 2+的应用。
更新日期:2018-01-16
中文翻译:
跨越有机超强碱应用的分子柔性的策略设计和利用:DFT研究
密度泛函理论(DFT)计算是针对一系列可能用作有机超碱的新型分子框架进行的。在本报告中,我们利用潜在的锚定基团1,5,7-三氮杂双环[4.4.0] dec-5-ene(TBD)开发并战略性地取代了吡啶的2和6位。计算了气相以及乙腈溶液中分子骨架的质子亲和力(PAs)值。发现设计的非离子和中性有机超碱具有比基准分子1,8-双(二甲基氨基)-萘(DMAN)更高的碱度。在理论水平为B3LYP / 6-311 + G ** // B3LYP / 6-31 + G *的情况下,针对设计的分子框架计算了零点振动能(ZPVE)和PAs值。对于压缩,我们还在M06-2X / 6-311 + G ** // M06-2X / 6-31 + G *理论水平上对所有结构进行了优化。分子框架图9在气相和乙腈溶液中的理论PA值分别最高为1151.2和1246.3 kJ mol -1,分别为B3LYP / 6-311 + G ** // B3LYP / 6-31 + G *。设计的分子框架具有更好的柔韧性,使其能够根据各种应用的需要,通过改变腔体的大小来选择性地提取较小至较大尺寸的分子。在这种情况下,我们探索了设计的分子框架在VO 2 +上选择性萃取UO 2 2+的应用。










































 京公网安备 11010802027423号
京公网安备 11010802027423号