European Journal of Medicinal Chemistry ( IF 6.0 ) Pub Date : 2018-01-11 , DOI: 10.1016/j.ejmech.2018.01.027 Rui Hu , Jie Gao , Rushangul Rozimamat , Haji Akber Aisa
|
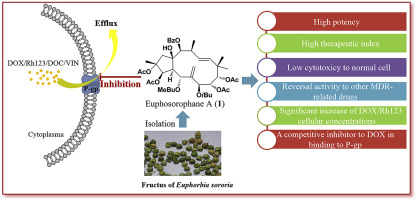
Five new (1–5) and ten known (6–15) jatrophane diterpenoids were isolated from the fructus of Euphorbia sororia and their structures were elucidated by extensive spectroscopic analysis. The absolute configurations of compounds 1 and 4 were confirmed by X-ray crystallographic analysis. Cytotoxicity and anti-multidrug resistance effects of these jatrophane diterpenoids were evaluated in multidrug-resistant MCF-7/ADR breast cancer cells with an overexpression of P-glycoprotein (P-gp). Eight compounds (1, 2, 4, 6, 8, 10, 11, and 15) showed promising chemoreversal abilities compared to verapamil (VRP). The most potent compound, Euphosorophane A (1), possessed many advantages, including (1) high potency (EC50 = 92.68 ± 18.28 nM) in reversing P-gp-mediated resistance to doxorubicin (DOX), low cytotoxicity, and a high therapeutic index, (2) potency in reversing resistance to other cytotoxic agents associated with MDR, and (3) inhibition of P-gp-mediated Rhodamine123 (Rh123) efflux function in MCF-7/ADR cells. The results of the Western blot analysis indicated that the multidrug resistance (MDR) reversal induced by 1 was not due to the inhibiton of P-gp expression. Compound 1 stimulated P-gp-ATPase activity and caused the dose-dependent inhibition of DOX transport activity. Lineweaver-Burk and Dixon plots implied that 1 was a competitive inhibitor to DOX in the binding site of P-gp with a Ki of 0.49–0.50 μM. Our data suggested that 1 had a high binding affinity toward the DOX recognition site of P-gp. This resulted in inhibiting DOX transport, increasing intracellular DOX concentration, and finally resensitizing MCF-7/ADR to DOX. In addition, we discussed some added contents in the structure-activity relationship (SAR) of jatrophane diterpenoids.
中文翻译:
大戟属的麻疯树二萜类化合物作为针对基于P糖蛋白的多药耐药性的有效调节剂
五个新的(15)和10已知的(6 - 15)jatrophane二萜类化合物是从的果实分离大戟sororia和其结构经过广泛光谱分析阐明。通过X射线晶体学分析证实了化合物1和4的绝对构型。在具有P-糖蛋白(P-gp)过表达的多药耐药MCF-7 / ADR乳腺癌细胞中评估了这些麻风树素二萜类化合物的细胞毒性和抗多药耐药性。八种化合物(1,2,4,6,8,10,11和15)与维拉帕米(VRP)相比,显示出了有希望的化学逆转能力。最有效的化合物Euphosorophane A(1)具有许多优点,包括(1 )在逆转P-gp介导的对阿霉素(DOX)的抗性方面具有高效力(EC 50 = 92.68±18.28 nM),低细胞毒性和高毒性。治疗指标,(2)逆转对与MDR相关的其他细胞毒性药物的抵抗力,以及(3)抑制MCF-7 / ADR细胞中P-gp介导的Rhodamine123(Rh123)外排功能。Western印迹分析的结果表明1诱导的多药耐药性(MDR)逆转不是由于抑制P-gp表达。化合物1刺激P-gp-ATPase活性并引起DOX转运活性的剂量依赖性抑制。双倒数和狄克逊曲线暗示1是一个竞争性抑制剂以DOX在P-gp的具有0.49-0.50的Ki的结合位点 μ M.我们的数据表明,1有朝向P的DOX识别位点的高结合亲和力-gp。这导致抑制DOX转运,增加细胞内DOX浓度,并最终使MCF-7 / ADR对DOX敏感。此外,我们讨论了麻疯树二萜类化合物的构效关系(SAR)中的一些附加内容。










































 京公网安备 11010802027423号
京公网安备 11010802027423号