当前位置:
X-MOL 学术
›
J. Proteome Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Mining the Secretome of C2C12 Muscle Cells: Data Dependent Experimental Approach To Analyze Protein Secretion Using Label-Free Quantification and Peptide Based Analysis
Journal of Proteome Research ( IF 3.8 ) Pub Date : 2018-01-24 00:00:00 , DOI: 10.1021/acs.jproteome.7b00684 Leonie Grube 1 , Rafael Dellen 2, 3 , Fabian Kruse 1 , Holger Schwender 2, 3 , Kai Stühler 1, 4 , Gereon Poschmann 1
Journal of Proteome Research ( IF 3.8 ) Pub Date : 2018-01-24 00:00:00 , DOI: 10.1021/acs.jproteome.7b00684 Leonie Grube 1 , Rafael Dellen 2, 3 , Fabian Kruse 1 , Holger Schwender 2, 3 , Kai Stühler 1, 4 , Gereon Poschmann 1
Affiliation
|
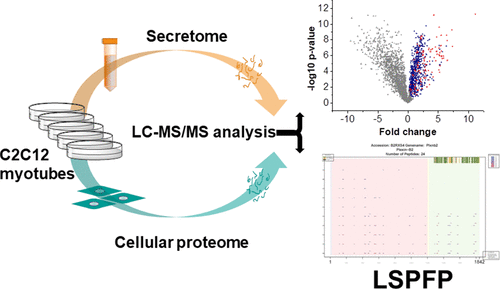
Secretome analysis faces several challenges including detection of low abundant proteins and the discrimination of bona fide secreted proteins from false-positive identifications stemming from cell leakage or serum. Here, we developed a two-step secretomics approach and applied it to the analysis of secreted proteins of C2C12 skeletal muscle cells since the skeletal muscle has been identified as an important endocrine organ secreting myokines as signaling molecules. First, we compared culture supernatants with corresponding cell lysates by mass spectrometry-based proteomics and label-free quantification. We identified 672 protein groups as candidate secreted proteins due to their higher abundance in the secretome. On the basis of Brefeldin A mediated blocking of classical secretory processes, we estimated a sensitivity of >80% for the detection of classical secreted proteins for our experimental approach. In the second step, the peptide level information was integrated with UniProt based protein information employing the newly developed bioinformatics tool “Lysate and Secretome Peptide Feature Plotter” (LSPFP) to detect proteolytic protein processing events that might occur during secretion. Concerning the proof of concept, we identified truncations of the cytoplasmic part of the protein Plexin-B2. Our workflow provides an efficient combination of experimental workflow and data analysis to identify putative secreted and proteolytic processed proteins.
中文翻译:

挖掘C2C12肌肉细胞的分泌组:使用无标记定量和基于肽的分析来分析蛋白质分泌的数据依赖实验方法
分泌组分析面临几个挑战,包括检测低丰度蛋白和区分真正分泌的蛋白,以及由于细胞渗漏或血清产生的假阳性鉴定结果。在这里,我们开发了一种两步分泌组学方法,并将其应用于C2C12骨骼肌细胞分泌蛋白的分析,因为骨骼肌已被确定为分泌肌动蛋白作为信号分子的重要内分泌器官。首先,我们通过基于质谱的蛋白质组学和无标记定量比较了培养物上清液和相应的细胞裂解液。我们将672个蛋白质组鉴定为候选分泌蛋白,因为它们在分泌组中的丰度更高。在布雷菲德菌素A介导的经典分泌过程阻滞的基础上,我们估计敏感性> 80%用于检测我们实验方法中的经典分泌蛋白。第二步,使用新开发的生物信息学工具“ Lysate and Secretome肽特征绘图仪”(LSPFP)将肽水平信息与基于UniProt的蛋白质信息整合在一起,以检测分泌过程中可能发生的蛋白水解蛋白加工事件。关于概念证明,我们鉴定了蛋白质Plexin-B2的胞质部分的截短。我们的工作流程提供了实验工作流程和数据分析的有效组合,以识别推定的分泌蛋白和蛋白水解加工蛋白。使用新开发的生物信息学工具“ Lysate and Secretome肽特征绘图仪”(LSPFP)将肽水平信息与基于UniProt的蛋白质信息集成在一起,以检测在分泌过程中可能发生的蛋白水解蛋白加工事件。关于概念证明,我们鉴定了蛋白质Plexin-B2的胞质部分的截短。我们的工作流程提供了实验工作流程和数据分析的有效组合,以识别推定的分泌蛋白和蛋白水解加工蛋白。使用新开发的生物信息学工具“ Lysate and Secretome肽特征绘图仪”(LSPFP)将肽水平信息与基于UniProt的蛋白质信息集成在一起,以检测在分泌过程中可能发生的蛋白水解蛋白加工事件。关于概念证明,我们鉴定了蛋白质Plexin-B2的胞质部分的截短。我们的工作流程提供了实验工作流程和数据分析的有效组合,以识别推定的分泌蛋白和蛋白水解加工蛋白。
更新日期:2018-01-25
中文翻译:
挖掘C2C12肌肉细胞的分泌组:使用无标记定量和基于肽的分析来分析蛋白质分泌的数据依赖实验方法
分泌组分析面临几个挑战,包括检测低丰度蛋白和区分真正分泌的蛋白,以及由于细胞渗漏或血清产生的假阳性鉴定结果。在这里,我们开发了一种两步分泌组学方法,并将其应用于C2C12骨骼肌细胞分泌蛋白的分析,因为骨骼肌已被确定为分泌肌动蛋白作为信号分子的重要内分泌器官。首先,我们通过基于质谱的蛋白质组学和无标记定量比较了培养物上清液和相应的细胞裂解液。我们将672个蛋白质组鉴定为候选分泌蛋白,因为它们在分泌组中的丰度更高。在布雷菲德菌素A介导的经典分泌过程阻滞的基础上,我们估计敏感性> 80%用于检测我们实验方法中的经典分泌蛋白。第二步,使用新开发的生物信息学工具“ Lysate and Secretome肽特征绘图仪”(LSPFP)将肽水平信息与基于UniProt的蛋白质信息整合在一起,以检测分泌过程中可能发生的蛋白水解蛋白加工事件。关于概念证明,我们鉴定了蛋白质Plexin-B2的胞质部分的截短。我们的工作流程提供了实验工作流程和数据分析的有效组合,以识别推定的分泌蛋白和蛋白水解加工蛋白。使用新开发的生物信息学工具“ Lysate and Secretome肽特征绘图仪”(LSPFP)将肽水平信息与基于UniProt的蛋白质信息集成在一起,以检测在分泌过程中可能发生的蛋白水解蛋白加工事件。关于概念证明,我们鉴定了蛋白质Plexin-B2的胞质部分的截短。我们的工作流程提供了实验工作流程和数据分析的有效组合,以识别推定的分泌蛋白和蛋白水解加工蛋白。使用新开发的生物信息学工具“ Lysate and Secretome肽特征绘图仪”(LSPFP)将肽水平信息与基于UniProt的蛋白质信息集成在一起,以检测在分泌过程中可能发生的蛋白水解蛋白加工事件。关于概念证明,我们鉴定了蛋白质Plexin-B2的胞质部分的截短。我们的工作流程提供了实验工作流程和数据分析的有效组合,以识别推定的分泌蛋白和蛋白水解加工蛋白。










































 京公网安备 11010802027423号
京公网安备 11010802027423号