当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
The ortho-benzyne cation is not planar†
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2018-01-09 00:00:00 , DOI: 10.1039/c7cp08055g D. Kaiser 1, 2, 3, 4 , E. Reusch 1, 2, 3, 4 , P. Hemberger 5, 6, 7, 8 , A. Bodi 5, 6, 7, 8 , E. Welz 1, 2, 3, 4 , B. Engels 1, 2, 3, 4 , I. Fischer 1, 2, 3, 4
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2018-01-09 00:00:00 , DOI: 10.1039/c7cp08055g D. Kaiser 1, 2, 3, 4 , E. Reusch 1, 2, 3, 4 , P. Hemberger 5, 6, 7, 8 , A. Bodi 5, 6, 7, 8 , E. Welz 1, 2, 3, 4 , B. Engels 1, 2, 3, 4 , I. Fischer 1, 2, 3, 4
Affiliation
|
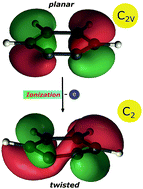
A recent review on the photoionisation of the C6H4 isomer ortho-benzyne suggests that bands reported in earlier photoelectron spectra might be due to side products or contaminations, while computations raise doubts, whether the cation has a planar geometry. We therefore reinvestigate the photoionisation of ortho-benzyne, generated by pyrolysis from benzocyclobutenedione, by photoion mass-selected threshold photoelectron (ms-TPE) spectroscopy using synchrotron radiation. The experiments are accompanied by a theoretical study that investigates the structure of the ortho-benzyne cation systematically as a function of the computational method, up to CASPT2(11,14) ab initio computations. Our study leads to a re-evaluation of the ionisation energy of ortho-benzyne. It reveals that the ortho-benzyne cation has indeed a twisted C2 geometry rather than a C2v structure. A vertical ionisation energy IEvert of 9.77 eV and an adiabatic ionisation energy of IEad = 9.56 eV are computed for ortho-benzyne. A Franck–Condon simulation of the photoelectron spectrum based on the CASPT2 results and including three electronic states of the cation is in agreement with the experiment and yields IEad = 9.51 eV (+50 meV/−100 meV). Since this value is in contrast with previous work, the ionisation energy has to be revised based on our study. Computational methods based on density functional theory give a reasonable description of the cationic ground state, but fail for the corresponding excited electronic states that are indispensible for a proper assignment of the photoelectron spectrum.
中文翻译:

该邻-benzyne阳离子不是平面†
最近对C 6 H 4异构体邻苯甲酰的光电离的评论表明,较早的光电子谱中报道的谱带可能是由于副产物或污染引起的,而计算却引起了人们对阳离子是否具有平面几何形状的疑问。因此,我们通过使用同步加速器辐射的光子质量选择阈值光电子(ms-TPE)光谱学,重新研究了由苯并环丁二酮热解生成的邻苯甲酰的光电离。实验伴随着一项理论研究,根据计算方法系统地研究了邻-苄基阳离子的结构,从头算到CASPT2(11,14)计算。我们的研究导致对邻-苯并ne的电离能的重新评估。它揭示了邻-苯甲酰基阳离子确实具有扭曲的C 2几何形状而不是C 2v结构。计算邻苯并苯的垂直电离能IE vert为9.77 eV,绝热电离能IE ad = 9.56 eV 。基于CASPT2结果并包括阳离子的三个电子状态的光电子谱的Franck-Condon模拟与实验相符,并产生IE ad= 9.51 eV(+50 meV / −100 meV)。由于该值与以前的工作相反,因此必须根据我们的研究来修改电离能。基于密度泛函理论的计算方法对阳离子基态进行了合理的描述,但是对于相应的激发电子态却是失败的,而对于适当分配光电子谱而言,该激发态是必不可少的。
更新日期:2018-01-09
中文翻译:
该邻-benzyne阳离子不是平面†
最近对C 6 H 4异构体邻苯甲酰的光电离的评论表明,较早的光电子谱中报道的谱带可能是由于副产物或污染引起的,而计算却引起了人们对阳离子是否具有平面几何形状的疑问。因此,我们通过使用同步加速器辐射的光子质量选择阈值光电子(ms-TPE)光谱学,重新研究了由苯并环丁二酮热解生成的邻苯甲酰的光电离。实验伴随着一项理论研究,根据计算方法系统地研究了邻-苄基阳离子的结构,从头算到CASPT2(11,14)计算。我们的研究导致对邻-苯并ne的电离能的重新评估。它揭示了邻-苯甲酰基阳离子确实具有扭曲的C 2几何形状而不是C 2v结构。计算邻苯并苯的垂直电离能IE vert为9.77 eV,绝热电离能IE ad = 9.56 eV 。基于CASPT2结果并包括阳离子的三个电子状态的光电子谱的Franck-Condon模拟与实验相符,并产生IE ad= 9.51 eV(+50 meV / −100 meV)。由于该值与以前的工作相反,因此必须根据我们的研究来修改电离能。基于密度泛函理论的计算方法对阳离子基态进行了合理的描述,但是对于相应的激发电子态却是失败的,而对于适当分配光电子谱而言,该激发态是必不可少的。










































 京公网安备 11010802027423号
京公网安备 11010802027423号