当前位置:
X-MOL 学术
›
Macromolecules
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Quantitative Comparison of Atomistic Simulations with Experiment for a Cross-Linked Epoxy: A Specific Volume–Cooling Rate Analysis
Macromolecules ( IF 5.1 ) Pub Date : 2018-01-08 00:00:00 , DOI: 10.1021/acs.macromol.7b01303 Ketan S. Khare 1, 2 , Frederick R. Phelan 2
Macromolecules ( IF 5.1 ) Pub Date : 2018-01-08 00:00:00 , DOI: 10.1021/acs.macromol.7b01303 Ketan S. Khare 1, 2 , Frederick R. Phelan 2
Affiliation
|
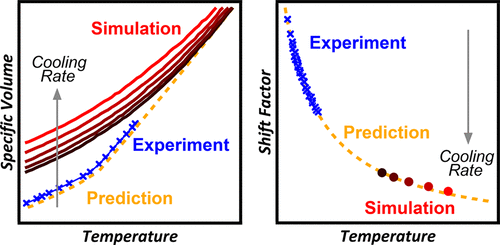
Cross-linked epoxy thermosets, like all glass-forming viscoelastic materials, show both a temperature and rate dependence in their thermomechanical properties. However, accounting for rate effects on these properties using molecular dynamics (MD) simulations and making quantitative comparison with experimental measurements has proven to be a difficult task due to the extreme mismatch between experimental and computationally accessible cooling rates. For this reason, the effect of cooling rate on material properties in glass-forming systems (including epoxy networks) has been mostly ignored in computational studies, making quantitative comparison with experimental data nebulous. In this work, we investigate a strategy for modeling rate effects in an epoxy network based on an approach that uses theoretically informed simulation and analysis protocols in combination with material specific time–temperature superposition (TTSP) data obtained from experimental measurements. To illustrate and test the strategy, we build and study an atomistic model of a cross-linked epoxy network. Molecular dynamics simulations are used to model the specific volume as a function of temperature across the glass transition from the rubbery to the glassy state using a total of five computationally accessible cooling rates. From the trends thus identified, we pinpoint the temperatures at which the models show rubbery and glassy behavior and use this information to calculate the values of the glass transition temperature (Tg) for each of the different cooling rates. Comparison with experimental data obtained from the literature (for the identical epoxy network) shows that our computations successfully predict the trends in specific volume in the rubbery and the glassy regions within 0.5%. We then compare the Tg values obtained from the data analysis with those calculated using the TTSP data obtained from the literature. Excellent agreement is found, and the Tg values from the two different methods are within 1.5% for all cooling rates. While our MD simulations do not replicate the experimental cooling rates, the agreement between these two sets of Tg values quantitatively relates the computational and experimental data sets. This agreement indicates that atomistic simulations can reliably capture the molecular mechanisms underlying viscoelasticity in this cross-linked epoxy (even when cooling rates differ by orders of magnitude) and that volume–rate analysis in conjunction with TTSP is a reliable method to computationally characterize certain classes of glass-forming materials. We believe that the general paradigm and protocols developed in this work should in principle be extensible as an efficacious means to integrate theory, computation, and experiment for the analysis of amorphous macromolecular materials.
中文翻译:

与交联环氧树脂实验的原子模拟定量比较:特定的体积冷却速率分析
像所有形成玻璃的粘弹性材料一样,交联的环氧热固性材料在其热机械性能中表现出温度和速率的依赖性。但是,由于实验和计算上可达到的冷却速率之间存在极大的不匹配,因此使用分子动力学(MD)模拟考虑速率对这些性能的影响并与实验测量值进行定量比较已被证明是一项艰巨的任务。因此,在计算研究中,冷却速率对玻璃成形系统(包括环氧网络)中材料性能的影响已被忽略,从而使与实验数据的定量比较模糊。在这项工作中,我们研究了一种在环氧树脂网络中建模速率效应的策略,该策略基于一种方法,该方法使用理论上已知的仿真和分析协议,并结合从实验测量中获得的材料特定的时温叠加(TTSP)数据。为了说明和测试该策略,我们建立并研究了交联环氧网络的原子模型。使用分子动力学模拟,使用总共五种可计算的冷却速率,将比体积与从橡胶态到玻璃态的玻璃化转变温度之间的函数关系进行建模。根据由此确定的趋势,我们可以精确地确定模型显示出橡胶态和玻璃态行为的温度,并使用此信息来计算玻璃化转变温度的值(为了说明和测试该策略,我们建立并研究了交联环氧网络的原子模型。使用分子动力学模拟,使用总共五种可计算的冷却速率,将比体积与从橡胶态到玻璃态的玻璃化转变温度之间的函数关系进行建模。根据由此确定的趋势,我们可以精确地确定模型显示出橡胶态和玻璃态行为的温度,并使用此信息来计算玻璃化转变温度的值(为了说明和测试该策略,我们建立并研究了交联环氧网络的原子模型。使用分子动力学模拟,使用总共五种可计算的冷却速率,将比体积与从橡胶态到玻璃态的玻璃化转变温度之间的函数关系进行建模。根据由此确定的趋势,我们可以精确地确定模型显示出橡胶态和玻璃态行为的温度,并使用此信息来计算玻璃化转变温度的值(对于每个不同的冷却速率,T g)。与从文献中获得的实验数据(对于相同的环氧网络)进行比较表明,我们的计算成功地预测了橡胶状区域和玻璃状区域中0.5%以内的比容趋势。然后,我们将从数据分析中获得的T g值与使用从文献中获得的TTSP数据计算出的T g值进行比较。发现极好的一致性,并且对于所有冷却速率,两种不同方法的T g值均在1.5%以内。尽管我们的MD模拟不能复制实验冷却速率,但这两套T g之间的一致性值定量地关联了计算和实验数据集。该协议表明,原子模拟可以可靠地捕获这种交联环氧树脂中粘弹性的分子机制(即使冷却速率相差一个数量级),并且体积速率分析与TTSP结合使用是一种可靠地计算某些类别的特征的方法玻璃形成材料。我们认为,这项工作中开发的一般范式和协议原则上应该是可扩展的,可以作为一种有效的手段来整合理论,计算和实验,以分析非晶态大分子材料。
更新日期:2018-01-08
中文翻译:
与交联环氧树脂实验的原子模拟定量比较:特定的体积冷却速率分析
像所有形成玻璃的粘弹性材料一样,交联的环氧热固性材料在其热机械性能中表现出温度和速率的依赖性。但是,由于实验和计算上可达到的冷却速率之间存在极大的不匹配,因此使用分子动力学(MD)模拟考虑速率对这些性能的影响并与实验测量值进行定量比较已被证明是一项艰巨的任务。因此,在计算研究中,冷却速率对玻璃成形系统(包括环氧网络)中材料性能的影响已被忽略,从而使与实验数据的定量比较模糊。在这项工作中,我们研究了一种在环氧树脂网络中建模速率效应的策略,该策略基于一种方法,该方法使用理论上已知的仿真和分析协议,并结合从实验测量中获得的材料特定的时温叠加(TTSP)数据。为了说明和测试该策略,我们建立并研究了交联环氧网络的原子模型。使用分子动力学模拟,使用总共五种可计算的冷却速率,将比体积与从橡胶态到玻璃态的玻璃化转变温度之间的函数关系进行建模。根据由此确定的趋势,我们可以精确地确定模型显示出橡胶态和玻璃态行为的温度,并使用此信息来计算玻璃化转变温度的值(为了说明和测试该策略,我们建立并研究了交联环氧网络的原子模型。使用分子动力学模拟,使用总共五种可计算的冷却速率,将比体积与从橡胶态到玻璃态的玻璃化转变温度之间的函数关系进行建模。根据由此确定的趋势,我们可以精确地确定模型显示出橡胶态和玻璃态行为的温度,并使用此信息来计算玻璃化转变温度的值(为了说明和测试该策略,我们建立并研究了交联环氧网络的原子模型。使用分子动力学模拟,使用总共五种可计算的冷却速率,将比体积与从橡胶态到玻璃态的玻璃化转变温度之间的函数关系进行建模。根据由此确定的趋势,我们可以精确地确定模型显示出橡胶态和玻璃态行为的温度,并使用此信息来计算玻璃化转变温度的值(对于每个不同的冷却速率,T g)。与从文献中获得的实验数据(对于相同的环氧网络)进行比较表明,我们的计算成功地预测了橡胶状区域和玻璃状区域中0.5%以内的比容趋势。然后,我们将从数据分析中获得的T g值与使用从文献中获得的TTSP数据计算出的T g值进行比较。发现极好的一致性,并且对于所有冷却速率,两种不同方法的T g值均在1.5%以内。尽管我们的MD模拟不能复制实验冷却速率,但这两套T g之间的一致性值定量地关联了计算和实验数据集。该协议表明,原子模拟可以可靠地捕获这种交联环氧树脂中粘弹性的分子机制(即使冷却速率相差一个数量级),并且体积速率分析与TTSP结合使用是一种可靠地计算某些类别的特征的方法玻璃形成材料。我们认为,这项工作中开发的一般范式和协议原则上应该是可扩展的,可以作为一种有效的手段来整合理论,计算和实验,以分析非晶态大分子材料。










































 京公网安备 11010802027423号
京公网安备 11010802027423号