当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Full Enzyme Complex Simulation: Interactions in Human Pyruvate Dehydrogenase Complex
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2018-01-24 00:00:00 , DOI: 10.1021/acs.jcim.7b00557 Samira Hezaveh 1 , An-Ping Zeng 1 , Uwe Jandt 1
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2018-01-24 00:00:00 , DOI: 10.1021/acs.jcim.7b00557 Samira Hezaveh 1 , An-Ping Zeng 1 , Uwe Jandt 1
Affiliation
|
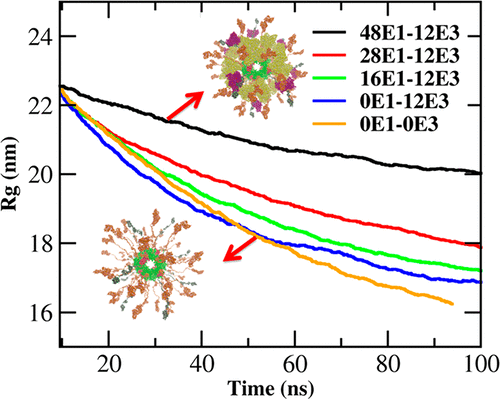
The pyruvate dehydrogenase complex (PDC) is a large macromolecular machine consisting of dozens of interacting enzymes that are connected and regulated by highly flexible domains, also called swinging arms. The overall structure and function of these domains and how they organize the complex function have not been elucidated in detail to date. This lack of structural and dynamic understanding is frequently observed in multidomain enzymatic complexes. Here we present the first full and dynamic structural model of full human PDC (hPDC), including binding of the linking arms to the surrounding E1 and E3 enzymes via their binding domains with variable stoichiometries. All of the linking domains were modeled at atomistic and coarse-grained levels, and the latter was parametrized to reproduce the same properties of those from the atomistic model. The radii of gyration of the wild-type full complex and functional trimeric subunits were in agreement with available experimental data. Furthermore, the E1 and E3 population effect on the overall structure of the full complex was studied. The results indicated that decreasing the number of E1s increases the flexibility of the now nonoccupied arms. Furthermore, their flexibility depends on the presence of other E1s and E3s in the vicinity, even if they are associated with other arms. As one consequence, the radius of gyration decreases with decreasing number of E1s. This effect also provides an indication of the optimal configuration of E1 and E3 on the basis of the assumption that a certain stability of the enymatic cloud is necessary to avoid free metabolic diffusion of intermediates (metabolic channeling). Our approach and results open a window for future enzyme engineering in a more effective way by evaluating the effect of different linker arm lengths, flexibilities, and combinations of mutations on the activity of other complex enzymes that involve flexible domains, including for example processive enzymes.
中文翻译:

完整的酶复合物模拟:人类丙酮酸脱氢酶复合物的相互作用
丙酮酸脱氢酶复合物(PDC)是一种大型的大分子机器,由数十种相互作用的酶组成,这些酶通过高度灵活的结构域(也称为摆动臂)连接并受其调节。迄今为止,这些域的整体结构和功能以及它们如何组织复杂的功能尚未得到详细阐明。在多域酶复合物中经常观察到这种缺乏结构和动态理解的现象。在这里,我们介绍了完整的人类PDC(hPDC)的第一个完整的动态结构模型,包括通过具有可变化学计量比的结合域将连接臂与周围的E1和E3酶结合。所有连接域都在原子级和粗粒度级建模,并且对后者进行了参数化设置,以再现与原子级模型相同的属性。野生型完整复合物和功能性三聚体亚基的回转半径与可获得的实验数据一致。此外,研究了E1和E3种群对完整复合体整体结构的影响。结果表明,减少E1的数量可以增加现在空着的武器的灵活性。此外,它们的灵活性取决于附近是否存在其他E1和E3,即使它们与其他手臂相关联也是如此。结果,回转半径随着E1数量的减少而减小。根据以下假设,该效果还为E1和E3的最佳构型提供了指示:酶云必须具有一定的稳定性,以避免中间体的自由代谢扩散(代谢通道)。
更新日期:2018-01-24
中文翻译:
完整的酶复合物模拟:人类丙酮酸脱氢酶复合物的相互作用
丙酮酸脱氢酶复合物(PDC)是一种大型的大分子机器,由数十种相互作用的酶组成,这些酶通过高度灵活的结构域(也称为摆动臂)连接并受其调节。迄今为止,这些域的整体结构和功能以及它们如何组织复杂的功能尚未得到详细阐明。在多域酶复合物中经常观察到这种缺乏结构和动态理解的现象。在这里,我们介绍了完整的人类PDC(hPDC)的第一个完整的动态结构模型,包括通过具有可变化学计量比的结合域将连接臂与周围的E1和E3酶结合。所有连接域都在原子级和粗粒度级建模,并且对后者进行了参数化设置,以再现与原子级模型相同的属性。野生型完整复合物和功能性三聚体亚基的回转半径与可获得的实验数据一致。此外,研究了E1和E3种群对完整复合体整体结构的影响。结果表明,减少E1的数量可以增加现在空着的武器的灵活性。此外,它们的灵活性取决于附近是否存在其他E1和E3,即使它们与其他手臂相关联也是如此。结果,回转半径随着E1数量的减少而减小。根据以下假设,该效果还为E1和E3的最佳构型提供了指示:酶云必须具有一定的稳定性,以避免中间体的自由代谢扩散(代谢通道)。










































 京公网安备 11010802027423号
京公网安备 11010802027423号