当前位置:
X-MOL 学术
›
J. Phys. Chem. Lett.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Full Quantum Dynamics Simulation of a Realistic Molecular System Using the Adaptive Time-Dependent Density Matrix Renormalization Group Method
The Journal of Physical Chemistry Letters ( IF 4.8 ) Pub Date : 2018-01-11 00:00:00 , DOI: 10.1021/acs.jpclett.7b03224 Yao Yao 1 , Ke-Wei Sun 2 , Zhen Luo 3 , Haibo Ma 3
The Journal of Physical Chemistry Letters ( IF 4.8 ) Pub Date : 2018-01-11 00:00:00 , DOI: 10.1021/acs.jpclett.7b03224 Yao Yao 1 , Ke-Wei Sun 2 , Zhen Luo 3 , Haibo Ma 3
Affiliation
|
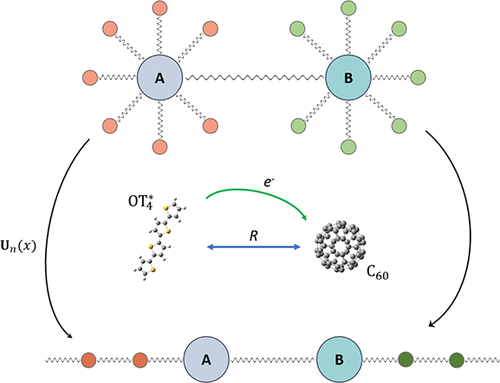
The accurate theoretical interpretation of ultrafast time-resolved spectroscopy experiments relies on full quantum dynamics simulations for the investigated system, which is nevertheless computationally prohibitive for realistic molecular systems with a large number of electronic and/or vibrational degrees of freedom. In this work, we propose a unitary transformation approach for realistic vibronic Hamiltonians, which can be coped with using the adaptive time-dependent density matrix renormalization group (t-DMRG) method to efficiently evolve the nonadiabatic dynamics of a large molecular system. We demonstrate the accuracy and efficiency of this approach with an example of simulating the exciton dissociation process within an oligothiophene/fullerene heterojunction, indicating that t-DMRG can be a promising method for full quantum dynamics simulation in large chemical systems. Moreover, it is also shown that the proper vibronic features in the ultrafast electronic process can be obtained by simulating the two-dimensional (2D) electronic spectrum by virtue of the high computational efficiency of the t-DMRG method.
中文翻译:

使用自适应时变密度矩阵重新归一化群方法的逼真的分子系统的全量子动力学模拟
超快速时间分辨光谱实验的准确理论解释依赖于所研究系统的完整量子动力学模拟,但对于具有大量电子和/或振动自由度的实际分子系统而言,这在计算上是令人望而却步的。在这项工作中,我们提出了一种用于现实振动哈密顿量的统一变换方法,该方法可以使用自适应时变密度矩阵重新归一化组(t-DMRG)方法来应对,以有效地演化大分子系统的非绝热动力学。我们以在寡噻吩/富勒烯异质结中模拟激子解离过程为例,证明了这种方法的准确性和效率,表明t-DMRG可能是在大型化学系统中进行全量子动力学模拟的有前途的方法。此外,还表明,借助于t-DMRG方法的高计算效率,可以通过模拟二维(2D)电子光谱来获得超快电子过程中适当的振动特征。
更新日期:2018-01-11
中文翻译:
使用自适应时变密度矩阵重新归一化群方法的逼真的分子系统的全量子动力学模拟
超快速时间分辨光谱实验的准确理论解释依赖于所研究系统的完整量子动力学模拟,但对于具有大量电子和/或振动自由度的实际分子系统而言,这在计算上是令人望而却步的。在这项工作中,我们提出了一种用于现实振动哈密顿量的统一变换方法,该方法可以使用自适应时变密度矩阵重新归一化组(t-DMRG)方法来应对,以有效地演化大分子系统的非绝热动力学。我们以在寡噻吩/富勒烯异质结中模拟激子解离过程为例,证明了这种方法的准确性和效率,表明t-DMRG可能是在大型化学系统中进行全量子动力学模拟的有前途的方法。此外,还表明,借助于t-DMRG方法的高计算效率,可以通过模拟二维(2D)电子光谱来获得超快电子过程中适当的振动特征。










































 京公网安备 11010802027423号
京公网安备 11010802027423号