当前位置:
X-MOL 学术
›
ACS Cent. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Learning a Local-Variable Model of Aromatic and Conjugated Systems
ACS Central Science ( IF 12.7 ) Pub Date : 2018-01-03 00:00:00 , DOI: 10.1021/acscentsci.7b00405 Matthew K. Matlock 1 , Na Le Dang 1 , S. Joshua Swamidass 1
ACS Central Science ( IF 12.7 ) Pub Date : 2018-01-03 00:00:00 , DOI: 10.1021/acscentsci.7b00405 Matthew K. Matlock 1 , Na Le Dang 1 , S. Joshua Swamidass 1
Affiliation
|
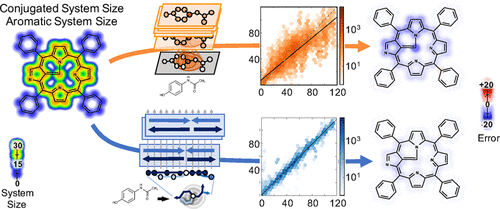
A collection of new approaches to building and training neural networks, collectively referred to as deep learning, are attracting attention in theoretical chemistry. Several groups aim to replace computationally expensive ab initio quantum mechanics calculations with learned estimators. This raises questions about the representability of complex quantum chemical systems with neural networks. Can local-variable models efficiently approximate nonlocal quantum chemical features? Here, we find that convolutional architectures, those that only aggregate information locally, cannot efficiently represent aromaticity and conjugation in large systems. They cannot represent long-range nonlocality known to be important in quantum chemistry. This study uses aromatic and conjugated systems computed from molecule graphs, though reproducing quantum simulations is the ultimate goal. This task, by definition, is both computable and known to be important to chemistry. The failure of convolutional architectures on this focused task calls into question their use in modeling quantum mechanics. To remedy this heretofore unrecognized deficiency, we introduce a new architecture that propagates information back and forth in waves of nonlinear computation. This architecture is still a local-variable model, and it is both computationally and representationally efficient, processing molecules in sublinear time with far fewer parameters than convolutional networks. Wave-like propagation models aromatic and conjugated systems with high accuracy, and even models the impact of small structural changes on large molecules. This new architecture demonstrates that some nonlocal features of quantum chemistry can be efficiently represented in local variable models.
中文翻译:

学习芳香和共轭系统的局部变量模型
建立和训练神经网络的新方法的集合(统称为深度学习)在理论化学中引起了人们的注意。几个小组旨在取代计算上昂贵的从头算用学得的估计量进行量子力学计算。这就提出了关于具有神经网络的复杂量子化学系统的可表示性的问题。局部变量模型可以有效地近似非局部量子化学特征吗?在这里,我们发现,仅在本地聚合信息的卷积架构无法有效地表示大型系统中的芳香性和共轭性。它们不能代表已知在量子化学中很重要的远程非局部性。尽管复制量子模拟是最终目标,但本研究使用从分子图计算出的芳香族和共轭体系。根据定义,该任务是可计算的,并且对化学很重要。卷积架构在此重点任务上的失败使人们质疑它们在量子力学建模中的用途。为了纠正迄今为止无法识别的缺陷,我们引入了一种新的体系结构,该体系结构在非线性计算浪潮中来回传播信息。该体系结构仍然是局部变量模型,并且在计算和表示效率上都非常有效,可以在亚线性时间内以比卷积网络少得多的参数来处理分子。波浪状传播可以对芳香族和共轭体系进行高精度建模,甚至可以模拟微小结构变化对大分子的影响。这种新的体系结构表明,可以在局部变量模型中有效地表示量子化学的某些非局部特征。该体系结构仍然是局部变量模型,并且在计算和表示效率上都非常有效,可以在亚线性时间内以比卷积网络少的参数处理分子。波浪状传播可以对芳香族和共轭体系进行高精度建模,甚至可以模拟微小结构变化对大分子的影响。这种新的体系结构表明,可以在局部变量模型中有效地表示量子化学的某些非局部特征。该体系结构仍然是局部变量模型,并且在计算和表示效率上都非常有效,可以在亚线性时间内以比卷积网络少得多的参数来处理分子。波浪状传播可以对芳香族和共轭体系进行高精度建模,甚至可以模拟微小结构变化对大分子的影响。这种新的体系结构表明,可以在局部变量模型中有效地表示量子化学的某些非局部特征。
更新日期:2018-01-03
中文翻译:
学习芳香和共轭系统的局部变量模型
建立和训练神经网络的新方法的集合(统称为深度学习)在理论化学中引起了人们的注意。几个小组旨在取代计算上昂贵的从头算用学得的估计量进行量子力学计算。这就提出了关于具有神经网络的复杂量子化学系统的可表示性的问题。局部变量模型可以有效地近似非局部量子化学特征吗?在这里,我们发现,仅在本地聚合信息的卷积架构无法有效地表示大型系统中的芳香性和共轭性。它们不能代表已知在量子化学中很重要的远程非局部性。尽管复制量子模拟是最终目标,但本研究使用从分子图计算出的芳香族和共轭体系。根据定义,该任务是可计算的,并且对化学很重要。卷积架构在此重点任务上的失败使人们质疑它们在量子力学建模中的用途。为了纠正迄今为止无法识别的缺陷,我们引入了一种新的体系结构,该体系结构在非线性计算浪潮中来回传播信息。该体系结构仍然是局部变量模型,并且在计算和表示效率上都非常有效,可以在亚线性时间内以比卷积网络少得多的参数来处理分子。波浪状传播可以对芳香族和共轭体系进行高精度建模,甚至可以模拟微小结构变化对大分子的影响。这种新的体系结构表明,可以在局部变量模型中有效地表示量子化学的某些非局部特征。该体系结构仍然是局部变量模型,并且在计算和表示效率上都非常有效,可以在亚线性时间内以比卷积网络少的参数处理分子。波浪状传播可以对芳香族和共轭体系进行高精度建模,甚至可以模拟微小结构变化对大分子的影响。这种新的体系结构表明,可以在局部变量模型中有效地表示量子化学的某些非局部特征。该体系结构仍然是局部变量模型,并且在计算和表示效率上都非常有效,可以在亚线性时间内以比卷积网络少得多的参数来处理分子。波浪状传播可以对芳香族和共轭体系进行高精度建模,甚至可以模拟微小结构变化对大分子的影响。这种新的体系结构表明,可以在局部变量模型中有效地表示量子化学的某些非局部特征。









































 京公网安备 11010802027423号
京公网安备 11010802027423号