当前位置:
X-MOL 学术
›
J. Phys. Chem. Lett.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Development of a Charge-Implicit ReaxFF Potential for Hydrocarbon Systems
The Journal of Physical Chemistry Letters ( IF 5.7 ) Pub Date : 2018-01-08 00:00:00 , DOI: 10.1021/acs.jpclett.7b03155 Michał Kański 1 , Dawid Maciążek 1 , Zbigniew Postawa 1 , Chowdhury M. Ashraf 2 , Adri C. T. van Duin 2 , Barbara J. Garrison 3
The Journal of Physical Chemistry Letters ( IF 5.7 ) Pub Date : 2018-01-08 00:00:00 , DOI: 10.1021/acs.jpclett.7b03155 Michał Kański 1 , Dawid Maciążek 1 , Zbigniew Postawa 1 , Chowdhury M. Ashraf 2 , Adri C. T. van Duin 2 , Barbara J. Garrison 3
Affiliation
|
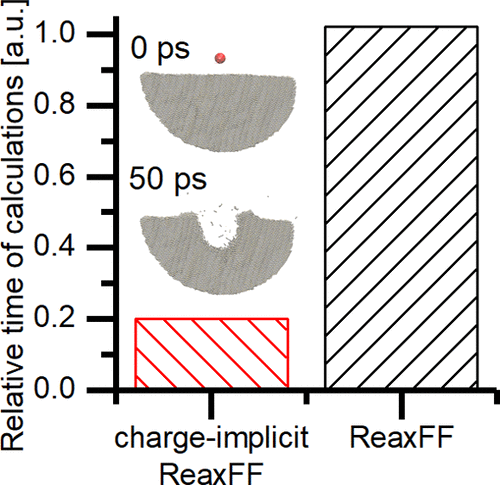
Molecular dynamics (MD) simulations continue to make important contributions to understanding chemical and physical processes. Concomitant with the growth of MD simulations is the need to have interaction potentials that both represent the chemistry of the system and are computationally efficient. We propose a modification to the ReaxFF potential for carbon and hydrogen that eliminates the time-consuming charge equilibration, eliminates the acknowledged flaws of the electronegativity equalization method, includes an expanded training set for condensed phases, has a repulsive wall for simulations of energetic particle bombardment, and is compatible with the LAMMPS code. This charge-implicit ReaxFF potential is five times faster than the conventional ReaxFF potential for a simulation of keV particle bombardment with a sample size of over 800 000 atoms.
中文翻译:

用于碳氢化合物系统的电荷隐式ReaxFF电位的开发
分子动力学(MD)模拟继续为理解化学和物理过程做出重要贡献。与MD模拟的发展相伴随的是,需要具有既代表系统化学性质又具有计算效率的相互作用潜能。我们建议对碳和氢的ReaxFF电位进行修改,以消除耗时的电荷平衡,消除电负性均衡方法的公认缺陷,包括针对凝聚相的扩展训练集,具有用于模拟高能粒子轰击的排斥壁,并且与LAMMPS代码兼容。
更新日期:2018-01-08
中文翻译:
用于碳氢化合物系统的电荷隐式ReaxFF电位的开发
分子动力学(MD)模拟继续为理解化学和物理过程做出重要贡献。与MD模拟的发展相伴随的是,需要具有既代表系统化学性质又具有计算效率的相互作用潜能。我们建议对碳和氢的ReaxFF电位进行修改,以消除耗时的电荷平衡,消除电负性均衡方法的公认缺陷,包括针对凝聚相的扩展训练集,具有用于模拟高能粒子轰击的排斥壁,并且与LAMMPS代码兼容。


























 京公网安备 11010802027423号
京公网安备 11010802027423号