当前位置:
X-MOL 学术
›
Anal. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
IMTBX and Grppr: Software for Top-Down Proteomics Utilizing Ion Mobility-Mass Spectrometry
Analytical Chemistry ( IF 6.7 ) Pub Date : 2018-01-16 00:00:00 , DOI: 10.1021/acs.analchem.7b04999 Dmitry M Avtonomov 1 , Daniel A Polasky 1 , Brandon T Ruotolo 1 , Alexey I Nesvizhskii 1
Analytical Chemistry ( IF 6.7 ) Pub Date : 2018-01-16 00:00:00 , DOI: 10.1021/acs.analchem.7b04999 Dmitry M Avtonomov 1 , Daniel A Polasky 1 , Brandon T Ruotolo 1 , Alexey I Nesvizhskii 1
Affiliation
|
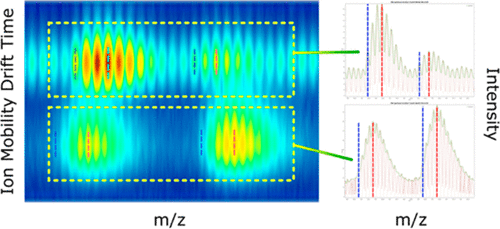
Top-down proteomics has emerged as a transformative method for the analysis of protein sequence and post-translational modifications (PTMs). Top-down experiments have historically been performed primarily on ultrahigh resolution mass spectrometers due to the complexity of spectra resulting from fragmentation of intact proteins, but recent advances in coupling ion mobility separations to faster, lower resolution mass analyzers now offer a viable alternative. However, software capable of interpreting the highly complex two-dimensional spectra that result from coupling ion mobility separation to top-down experiments is currently lacking. In this manuscript we present a software suite consisting of two programs, IMTBX (“IM Toolbox”) and Grppr (“Grouper”), that enable fully automated processing of such data. We demonstrate the capabilities of this software suite by examining a series of intact proteins on a Waters Synapt G2 ion-mobility equipped mass spectrometer and compare the results to the manual and semiautomated data analysis procedures we have used previously.
中文翻译:

IMTBX 和 Grppr:利用离子淌度质谱的自上而下蛋白质组学软件
自上而下的蛋白质组学已成为分析蛋白质序列和翻译后修饰 (PTM) 的革命性方法。由于完整蛋白质的碎片导致光谱的复杂性,自上而下的实验历来主要在超高分辨率质谱仪上进行,但将离子淌度分离与更快、分辨率较低的质量分析仪耦合的最新进展现在提供了一种可行的替代方案。然而,目前缺乏能够解释由离子淌度分离耦合到自上而下实验产生的高度复杂二维光谱的软件。在这篇手稿中,我们提出了一个由两个程序组成的软件套件:IMTBX(“IM Toolbox”)和 Grppr(“Grouper”),可以实现此类数据的全自动处理。我们通过在配备离子淌度的 Waters Synapt G2 质谱仪上检查一系列完整蛋白质,并将结果与我们之前使用的手动和半自动数据分析程序进行比较,展示了该软件套件的功能。
更新日期:2018-01-16
中文翻译:
IMTBX 和 Grppr:利用离子淌度质谱的自上而下蛋白质组学软件
自上而下的蛋白质组学已成为分析蛋白质序列和翻译后修饰 (PTM) 的革命性方法。由于完整蛋白质的碎片导致光谱的复杂性,自上而下的实验历来主要在超高分辨率质谱仪上进行,但将离子淌度分离与更快、分辨率较低的质量分析仪耦合的最新进展现在提供了一种可行的替代方案。然而,目前缺乏能够解释由离子淌度分离耦合到自上而下实验产生的高度复杂二维光谱的软件。在这篇手稿中,我们提出了一个由两个程序组成的软件套件:IMTBX(“IM Toolbox”)和 Grppr(“Grouper”),可以实现此类数据的全自动处理。我们通过在配备离子淌度的 Waters Synapt G2 质谱仪上检查一系列完整蛋白质,并将结果与我们之前使用的手动和半自动数据分析程序进行比较,展示了该软件套件的功能。










































 京公网安备 11010802027423号
京公网安备 11010802027423号