当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Saturation Mutagenesis by Efficient Free-Energy Calculation.
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2018-01-08 , DOI: 10.1021/acs.jctc.7b01099 Zuzana Jandova 1 , Daniel Fast 1 , Martina Setz 1 , Maria Pechlaner 1 , Chris Oostenbrink 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2018-01-08 , DOI: 10.1021/acs.jctc.7b01099 Zuzana Jandova 1 , Daniel Fast 1 , Martina Setz 1 , Maria Pechlaner 1 , Chris Oostenbrink 1
Affiliation
|
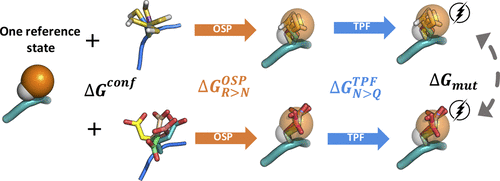
Single-point mutations in proteins can greatly influence protein stability, binding affinity, protein function or its expression per se. Here, we present accurate and efficient predictions of the free energy of mutation of amino acids. We divided the complete mutational free energy into an uncharging step, which we approximate by a third-power fitting (TPF) approach, and an annihilation step, which we approximate using the one-step perturbation (OSP) method. As a diverse set of test systems, we computed the solvation free energy of all amino acid side chain analogues and obtained an excellent agreement with thermodynamic integration (TI) data. Moreover, we calculated mutational free energies in model tripeptides and established an efficient protocol involving a single reference state. Again, the approximate methods agreed excellently with the TI references, with a root-mean-square error of only 3.6 kJ/mol over 17 mutations. Our combined TPF+OSP approach does show not only a very good agreement but also a 2-fold higher efficiency than full blown TI calculations.
中文翻译:

通过有效的自由能计算进行饱和诱变。
蛋白质中的单点突变可以极大地影响蛋白质稳定性、结合亲和力、蛋白质功能或其表达本身。在这里,我们对氨基酸突变的自由能进行了准确有效的预测。我们将完整的突变自由能分为一个不带电步骤,我们通过三次方拟合 (TPF) 方法来近似,以及一个湮灭步骤,我们使用一步扰动 (OSP) 方法来近似。作为一组多样化的测试系统,我们计算了所有氨基酸侧链类似物的溶剂化自由能,并与热力学积分 (TI) 数据获得了极好的一致性。此外,我们计算了模型三肽中的突变自由能,并建立了一个涉及单一参考状态的有效协议。再次,近似方法与 TI 参考文献非常吻合,17 个突变的均方根误差仅为 3.6 kJ/mol。我们的 TPF+OSP 组合方法不仅显示出非常好的一致性,而且效率比完整的 TI 计算高 2 倍。
更新日期:2018-01-08
中文翻译:
通过有效的自由能计算进行饱和诱变。
蛋白质中的单点突变可以极大地影响蛋白质稳定性、结合亲和力、蛋白质功能或其表达本身。在这里,我们对氨基酸突变的自由能进行了准确有效的预测。我们将完整的突变自由能分为一个不带电步骤,我们通过三次方拟合 (TPF) 方法来近似,以及一个湮灭步骤,我们使用一步扰动 (OSP) 方法来近似。作为一组多样化的测试系统,我们计算了所有氨基酸侧链类似物的溶剂化自由能,并与热力学积分 (TI) 数据获得了极好的一致性。此外,我们计算了模型三肽中的突变自由能,并建立了一个涉及单一参考状态的有效协议。再次,近似方法与 TI 参考文献非常吻合,17 个突变的均方根误差仅为 3.6 kJ/mol。我们的 TPF+OSP 组合方法不仅显示出非常好的一致性,而且效率比完整的 TI 计算高 2 倍。










































 京公网安备 11010802027423号
京公网安备 11010802027423号