当前位置:
X-MOL 学术
›
Chem. Bio. Drug Des.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Design, synthesis, and antiprotozoal evaluation of new 2,9‐bis[(substituted‐aminomethyl)phenyl]‐1,10‐phenanthroline derivatives
Chemical Biology & Drug Design ( IF 3.2 ) Pub Date : 2018-01-17 , DOI: 10.1111/cbdd.13164 Jean Guillon 1 , Anita Cohen 2 , Rabindra Nath Das 1 , Clotilde Boudot 3, 4 , Nassima Meriem Gueddouda 1 , Stéphane Moreau 1 , Luisa Ronga 1 , Solène Savrimoutou 1 , Louise Basmaciyan 2 , Camille Tisnerat 1 , Sacha Mestanier 1 , Sandra Rubio 1 , Sophia Amaziane 1 , Alexandra Dassonville-Klimpt 5 , Nadine Azas 2 , Bertrand Courtioux 3, 4 , Jean-Louis Mergny 1, 6 , Catherine Mullié 5 , Pascal Sonnet 5
Chemical Biology & Drug Design ( IF 3.2 ) Pub Date : 2018-01-17 , DOI: 10.1111/cbdd.13164 Jean Guillon 1 , Anita Cohen 2 , Rabindra Nath Das 1 , Clotilde Boudot 3, 4 , Nassima Meriem Gueddouda 1 , Stéphane Moreau 1 , Luisa Ronga 1 , Solène Savrimoutou 1 , Louise Basmaciyan 2 , Camille Tisnerat 1 , Sacha Mestanier 1 , Sandra Rubio 1 , Sophia Amaziane 1 , Alexandra Dassonville-Klimpt 5 , Nadine Azas 2 , Bertrand Courtioux 3, 4 , Jean-Louis Mergny 1, 6 , Catherine Mullié 5 , Pascal Sonnet 5
Affiliation
|
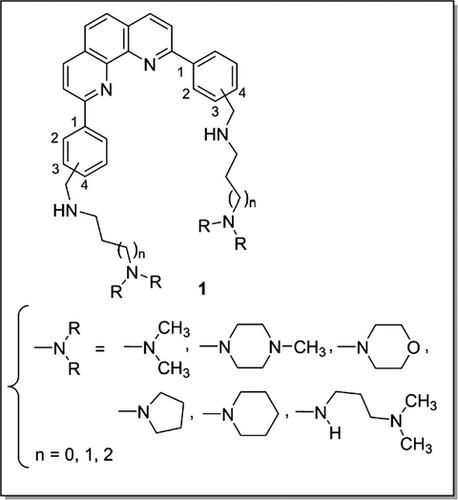
A series of new 2,9‐bis[(substituted‐aminomethyl)phenyl]‐1,10‐phenanthroline derivatives was synthesized, and the compounds were screened in vitro against three protozoan parasites (Plasmodium falciparum, Leishmania donovani, and Trypanosoma brucei brucei). Biological results showed antiparasitic activity with IC50 values in the μm range. The in vitro cytotoxicity of these molecules was assessed by incubation with human HepG2 cells; for some derivatives, cytotoxicity was observed at significantly higher concentrations than antiparasitic activity. The 2,9‐bis[(substituted‐aminomethyl)phenyl]‐1,10‐phenanthroline 1h was identified as the most potent antimalarial candidate with ratios of cytotoxic‐to‐antiparasitic activities of 107 and 39 against a chloroquine‐sensitive and a chloroquine‐resistant strain of P. falciparum, respectively. As the telomeres of the parasite P. falciparum are the likely target of this compound, we investigated stabilization of the Plasmodium telomeric G‐quadruplexes by our phenanthroline derivatives through a FRET melting assay. The ligands 1f and 1m were noticed to be more specific for FPf8T with higher stabilization for FPf8T than for the human F21T sequence.
中文翻译:

新的2,9-双[(取代-氨基甲基)苯基] -1,10-菲咯啉衍生物的设计,合成和抗原生动物评估
合成了一系列新的2,9-双[(取代-氨基甲基)苯基] -1,10-菲咯啉衍生物,并针对三种原生动物寄生虫(恶性疟原虫,利什曼原虫donovani和布鲁氏锥虫)对化合物进行了体外筛选。。生物学结果显示出抗寄生虫活性,IC 50值在μm范围内。这些分子的体外细胞毒性是通过与人类HepG2细胞孵育来评估的;对于某些衍生物,在比抗寄生虫活性高得多的浓度下观察到细胞毒性。2,9-双[(取代-氨基甲基)苯基] -1,10-菲咯啉1h被确定为最有效的抗疟疾候选药物,其对恶性疟原虫的氯喹敏感性和氯喹耐药性菌株的细胞毒/抗寄生虫活性分别为107和39 。由于恶性疟原虫的端粒是该化合物的靶标,因此我们通过FRET熔解法研究了菲咯啉衍生物对疟原虫端粒G-四链体的稳定作用。注意到配体1f和1m对FPf8T具有更高的特异性,对FPf8T具有比对人F21T序列更高的稳定性。
更新日期:2018-01-17
中文翻译:
新的2,9-双[(取代-氨基甲基)苯基] -1,10-菲咯啉衍生物的设计,合成和抗原生动物评估
合成了一系列新的2,9-双[(取代-氨基甲基)苯基] -1,10-菲咯啉衍生物,并针对三种原生动物寄生虫(恶性疟原虫,利什曼原虫donovani和布鲁氏锥虫)对化合物进行了体外筛选。。生物学结果显示出抗寄生虫活性,IC 50值在μm范围内。这些分子的体外细胞毒性是通过与人类HepG2细胞孵育来评估的;对于某些衍生物,在比抗寄生虫活性高得多的浓度下观察到细胞毒性。2,9-双[(取代-氨基甲基)苯基] -1,10-菲咯啉1h被确定为最有效的抗疟疾候选药物,其对恶性疟原虫的氯喹敏感性和氯喹耐药性菌株的细胞毒/抗寄生虫活性分别为107和39 。由于恶性疟原虫的端粒是该化合物的靶标,因此我们通过FRET熔解法研究了菲咯啉衍生物对疟原虫端粒G-四链体的稳定作用。注意到配体1f和1m对FPf8T具有更高的特异性,对FPf8T具有比对人F21T序列更高的稳定性。










































 京公网安备 11010802027423号
京公网安备 11010802027423号