当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Strategical Designing of Donor–Acceptor–Donor Based Organic Molecules for Tuning Their Linear Optical Properties
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2018-01-05 00:00:00 , DOI: 10.1021/acs.jpca.7b07381 Raja Sen 1 , Samarendra P. Singh 1 , Priya Johari 1
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2018-01-05 00:00:00 , DOI: 10.1021/acs.jpca.7b07381 Raja Sen 1 , Samarendra P. Singh 1 , Priya Johari 1
Affiliation
|
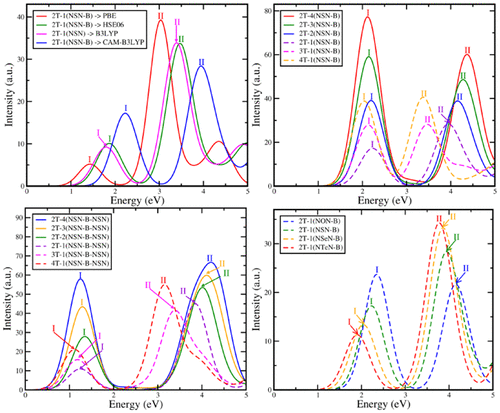
Low-energy linear absorption spectrum of a series of 48 donor–acceptor–donor (D–A–D) scheme based thiophone–benzo(bis-)X-diazole molecules with X = O, S, Se, or Te are calculated using time dependent density functional theory in order to propose strategical design of molecules that can efficiently absorb light in the infrared and visible region of the solar spectrum. Our study establishes that optical properties of the D–A–D based organic molecules significantly depend on the donor-to-acceptor (D/A) ratio and the strength of the acceptor moiety. Thus, by choice of a suitable D/A ratio and type of the acceptor moiety, the linear absorption spectrum can be largely shifted, in general, while the optical gap can be engineered over a wide energy range of ∼0.2–2.3 eV, in particular. It is also noticed that the increase in acceptor units (i.e., when D/A ≤ 1) leads to increase in steric hindrance in between them. This, in turn, disrupts the effective conjugation length and increases the optical gap. However, this effect is found to dominate strongly in the bis-configurations of the molecules as compared to the nonbis compositions. In order to reduce this effect for rational designing of effective D–A–D type chromophores with less steric hindrance, the role of π-conjugated ethylene (−CH═CH−) linkage/spacer between the A–A units is explored further. Here, it is found that introduction of such linkage substantially decreases the steric hindrance and, thereby, the optical gap as well. Besides this, our study also highlights and explains the impact of the acceptor moiety in improving the absorption capabilities of these molecules in the low-energy region.
中文翻译:

基于供体-受体-供体的有机分子的线性光学性质的策略设计
使用以下公式计算一系列基于48种供体-受体-供体(D–A–D)方案的硫磺-苯并(bis-)X-二唑分子的低能线性吸收光谱,其中X = O,S,Se或Te时变密度泛函理论,以便提出可以有效吸收太阳光谱的红外和可见光区域内分子的战略设计。我们的研究表明,基于D–A–D的有机分子的光学性质在很大程度上取决于供体与受体(D / A)的比例和受体部分的强度。因此,通常通过选择合适的D / A比和受体部分的类型,线性吸收光谱可以大大改变,而光学间隙可以在约0.2–2.3 eV的宽能量范围内进行工程设计,特别的。还应注意,受体单位的增加(即,当D / A≤1)导致它们之间的空间位阻增加。继而,这破坏了有效的共轭长度并增加了光学间隙。然而,与nonbis组合物相比,发现该作用在分子的双构型中起主要作用。为了减少这种影响,以合理设计有效的D–A–D型发色团,减少空间位阻,进一步探讨了A–A单元之间的π-共轭乙烯(-CH═CH-)键/间隔基的作用。在此发现,引入这种键合实质上减少了空间位阻,从而也减少了光学间隙。除此之外,我们的研究还突出并解释了受体部分对改善这些分子在低能区域中的吸收能力的影响。
更新日期:2018-01-05
中文翻译:
基于供体-受体-供体的有机分子的线性光学性质的策略设计
使用以下公式计算一系列基于48种供体-受体-供体(D–A–D)方案的硫磺-苯并(bis-)X-二唑分子的低能线性吸收光谱,其中X = O,S,Se或Te时变密度泛函理论,以便提出可以有效吸收太阳光谱的红外和可见光区域内分子的战略设计。我们的研究表明,基于D–A–D的有机分子的光学性质在很大程度上取决于供体与受体(D / A)的比例和受体部分的强度。因此,通常通过选择合适的D / A比和受体部分的类型,线性吸收光谱可以大大改变,而光学间隙可以在约0.2–2.3 eV的宽能量范围内进行工程设计,特别的。还应注意,受体单位的增加(即,当D / A≤1)导致它们之间的空间位阻增加。继而,这破坏了有效的共轭长度并增加了光学间隙。然而,与nonbis组合物相比,发现该作用在分子的双构型中起主要作用。为了减少这种影响,以合理设计有效的D–A–D型发色团,减少空间位阻,进一步探讨了A–A单元之间的π-共轭乙烯(-CH═CH-)键/间隔基的作用。在此发现,引入这种键合实质上减少了空间位阻,从而也减少了光学间隙。除此之外,我们的研究还突出并解释了受体部分对改善这些分子在低能区域中的吸收能力的影响。








































 京公网安备 11010802027423号
京公网安备 11010802027423号