当前位置:
X-MOL 学术
›
J. Med. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Structure-Based Design, Synthesis, and In Vivo Antinociceptive Effects of Selective A1 Adenosine Receptor Agonists
Journal of Medicinal Chemistry ( IF 6.8 ) Pub Date : 2018-01-03 00:00:00 , DOI: 10.1021/acs.jmedchem.7b01399 Riccardo Petrelli 1 , Mirko Scortichini 1 , Carmela Belardo 2 , Serena Boccella 2 , Livio Luongo 2 , Fabio Capone 3 , Sonja Kachler 4 , Patrizia Vita 1 , Fabio Del Bello 1 , Sabatino Maione 2 , Antonio Lavecchia 3 , Karl-Norbert Klotz 4 , Loredana Cappellacci 1
Journal of Medicinal Chemistry ( IF 6.8 ) Pub Date : 2018-01-03 00:00:00 , DOI: 10.1021/acs.jmedchem.7b01399 Riccardo Petrelli 1 , Mirko Scortichini 1 , Carmela Belardo 2 , Serena Boccella 2 , Livio Luongo 2 , Fabio Capone 3 , Sonja Kachler 4 , Patrizia Vita 1 , Fabio Del Bello 1 , Sabatino Maione 2 , Antonio Lavecchia 3 , Karl-Norbert Klotz 4 , Loredana Cappellacci 1
Affiliation
|
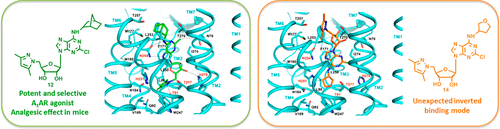
Our previous work discovered that combining the appropriate 5′- and N6-substitution in adenosine derivatives leads to the highly selective human A1 adenosine receptor (hA1AR) agonists or highly potent dual hA1AR agonists and hA3AR antagonists. In order to explore novel dual adenosine receptor ligands, a series of N6-substituted-5′-pyrazolyl-adenosine and 2-chloro-adenosine derivatives were synthesized and assayed in vitro at all ARs. The N6-(±)-endo-norbornyl derivative 12 was the most potent and selective at A1AR and effective as an analgesic in formalin test in mice, but none of the 5′-pyrazolyl series compounds showed a dual behavior at hA1 and hA3AR. Molecular modeling studies rationalized the structure–activity relationships and the selectivity profiles of the new series of A1AR agonists. Interestingly, an unexpected inverted binding mode of the N6-tetrahydrofuranyl derivative 14 was hypothesized to explain its low affinity at A1AR.
中文翻译:

选择性A 1腺苷受体激动剂的基于结构的设计,合成和体内抗伤害作用
我们以前的工作发现,在腺苷衍生物中结合适当的5'-和N 6-取代可导致高度选择性的人A 1腺苷受体(hA 1 AR)激动剂或高效的双重hA 1 AR激动剂和hA 3 AR拮抗剂。为了探索新颖的双腺苷受体配体,合成了一系列N 6-取代的5'-吡唑基-腺苷和2-氯-腺苷衍生物,并在所有AR中体外测定。所述Ñ 6 - (±) -内型-norbornyl衍生物12是最有效的和选择性A处1AR在福尔马林试验中可作为镇痛药有效,但5'-吡唑基系列化合物均未在hA 1和hA 3 AR处表现出双重行为。分子建模研究合理化了一系列新的A 1 AR激动剂的结构-活性关系和选择性。有趣的是,假设了N 6-四氢呋喃基衍生物14的意外的反向结合模式,以解释其对A 1 AR的低亲和力。
更新日期:2018-01-03
中文翻译:
选择性A 1腺苷受体激动剂的基于结构的设计,合成和体内抗伤害作用
我们以前的工作发现,在腺苷衍生物中结合适当的5'-和N 6-取代可导致高度选择性的人A 1腺苷受体(hA 1 AR)激动剂或高效的双重hA 1 AR激动剂和hA 3 AR拮抗剂。为了探索新颖的双腺苷受体配体,合成了一系列N 6-取代的5'-吡唑基-腺苷和2-氯-腺苷衍生物,并在所有AR中体外测定。所述Ñ 6 - (±) -内型-norbornyl衍生物12是最有效的和选择性A处1AR在福尔马林试验中可作为镇痛药有效,但5'-吡唑基系列化合物均未在hA 1和hA 3 AR处表现出双重行为。分子建模研究合理化了一系列新的A 1 AR激动剂的结构-活性关系和选择性。有趣的是,假设了N 6-四氢呋喃基衍生物14的意外的反向结合模式,以解释其对A 1 AR的低亲和力。










































 京公网安备 11010802027423号
京公网安备 11010802027423号