当前位置:
X-MOL 学术
›
J. Chem. Eng. Data
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Chain Length Dependence of the Thermodynamic Properties of n-Alkanes and their Monosubstituted Derivatives
Journal of Chemical & Engineering Data ( IF 2.0 ) Pub Date : 2017-12-19 00:00:00 , DOI: 10.1021/acs.jced.7b00837 José C. S. Costa 1, 2 , Adélio Mendes 2 , Luís M. N. B. F. Santos 1
Journal of Chemical & Engineering Data ( IF 2.0 ) Pub Date : 2017-12-19 00:00:00 , DOI: 10.1021/acs.jced.7b00837 José C. S. Costa 1, 2 , Adélio Mendes 2 , Luís M. N. B. F. Santos 1
Affiliation
|
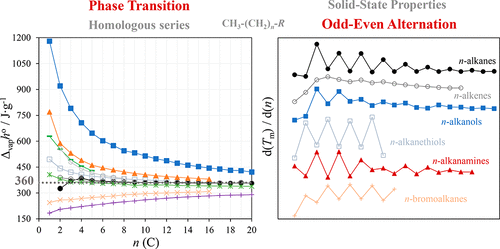
The present work presents an extensive literature survey and analysis of the heat capacity and thermodynamic properties of fusion, vaporization, and sublimation for the linear hydrocarbons and several terminally substituted homologous series. The successive introduction of methylene groups on the relative stability of the solid and liquid phases is analyzed and discussed based on the chain length dependence of the enthalpies, entropies, and Gibbs energies of phase transition. An odd–even alternation is observed in the fusion and sublimation equilibria. The improved packing patterns of even-numbered n-alkanes is reflected in higher values of melting temperatures and thermodynamic properties of phase transition. Molar heat capacities in liquid phase of n-alkanes derivatives exhibit a linear dependence with the chain length by an increment of 31 ± 2 J·K–1·mol–1 per methylene group (−CH2−). A contribution of 4.95 kJ·mol–1 per methylene group (value corrected for 298.15 K) is derived for the increment of the enthalpy of vaporization. A constant value for the specific enthalpy of vaporization is observed for long chain compounds: 360 J·g–1. As predictable, the enthalpy of vaporization is higher for groups that can form hydrogen-bonding interactions than for plain hydrocarbons. Concerning the monohalogenated alkanes, a clear increasing of enthalpy of vaporization for the larger halogen groups is observed. Moreover, the thermodynamic results indicate that along the fusion of n-alkanes and n-alkanols, there is a decrease of around 40% in the magnitude of intermolecular interactions.
中文翻译:

正构烷烃及其单取代衍生物热力学性质的链长依赖性
目前的工作提出了广泛的文献调查和分析的热容量和热力学性质的熔化,汽化和升华的线性碳氢化合物和几个末端取代的同源系列。基于相变的焓,熵和吉布斯能量的链长依赖性,对固相和液相相对稳定性上亚甲基的连续引入进行了分析和讨论。在融合和升华平衡中观察到奇偶交替。偶数正构烷烃的改进堆积模式反映在较高的熔点温度和相变的热力学性质上。液相的摩尔热容量n-烷烃衍生物与链长的线性关系呈线性关系,每亚甲基(-CH 2-)的增量为31±2 J·K –1 ·mol –1。每个亚甲基的贡献为4.95 kJ·mol –1(校正值为298.15 K),用于增加蒸发焓。对于长链化合物,观察到了比气化焓的恒定值:360 J·g –1。可以预见,可以形成氢键相互作用的基团的汽化焓要比普通烃更高。关于单卤代烷烃,观察到较大的卤素基团的汽化焓明显增加。此外,热力学结果表明,沿融合ñ -烷烃和Ñ链烷醇,有40%左右的分子间相互作用的大小的减小。
更新日期:2017-12-19
中文翻译:
正构烷烃及其单取代衍生物热力学性质的链长依赖性
目前的工作提出了广泛的文献调查和分析的热容量和热力学性质的熔化,汽化和升华的线性碳氢化合物和几个末端取代的同源系列。基于相变的焓,熵和吉布斯能量的链长依赖性,对固相和液相相对稳定性上亚甲基的连续引入进行了分析和讨论。在融合和升华平衡中观察到奇偶交替。偶数正构烷烃的改进堆积模式反映在较高的熔点温度和相变的热力学性质上。液相的摩尔热容量n-烷烃衍生物与链长的线性关系呈线性关系,每亚甲基(-CH 2-)的增量为31±2 J·K –1 ·mol –1。每个亚甲基的贡献为4.95 kJ·mol –1(校正值为298.15 K),用于增加蒸发焓。对于长链化合物,观察到了比气化焓的恒定值:360 J·g –1。可以预见,可以形成氢键相互作用的基团的汽化焓要比普通烃更高。关于单卤代烷烃,观察到较大的卤素基团的汽化焓明显增加。此外,热力学结果表明,沿融合ñ -烷烃和Ñ链烷醇,有40%左右的分子间相互作用的大小的减小。








































 京公网安备 11010802027423号
京公网安备 11010802027423号