当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Assessing the Molecular Basis of the Fuel Octane Scale: A Detailed Investigation on the Rate Controlling Steps of the Autoignition of Heptane and Isooctane
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2018-01-05 00:00:00 , DOI: 10.1021/acs.jpca.7b08521 João G. S. Monteiro 1 , André G. H. Barbosa 1 , André M. Henriques 1 , Pedro H. G. Neves 1 , Roberto S. Furtado 1 , Rodrigo M. Menezes 1 , Anderson R. dos Santos 2 , Felipe P. Fleming 2
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2018-01-05 00:00:00 , DOI: 10.1021/acs.jpca.7b08521 João G. S. Monteiro 1 , André G. H. Barbosa 1 , André M. Henriques 1 , Pedro H. G. Neves 1 , Roberto S. Furtado 1 , Rodrigo M. Menezes 1 , Anderson R. dos Santos 2 , Felipe P. Fleming 2
Affiliation
|
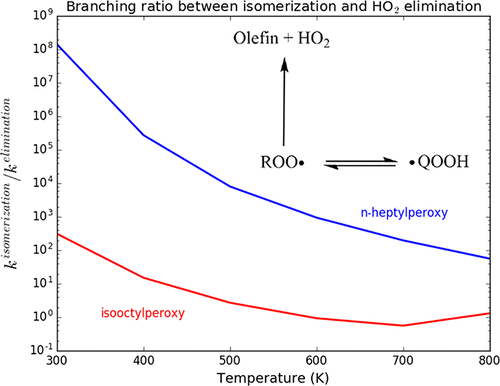
N-Heptane and 2,2,4-trimethylpentane (isooctane) are the key species in the modeling of ignition of hydrocarbon-based fuel formulations. Isooctane is knock-resistant whereas n-heptane is a very knock-prone hydrocarbon. It has been suggested that interconversion of their associated alkylperoxy and hydroperoxyalkyl species via hydrogen-transfer isomerization reaction is the key step to understand their different knocking behavior. In this work, the kinetics of unimolecular hydrogen-transfer reactions of n-heptylperoxy and isooctylperoxy are determined using canonical variational transition-state theory and multidimensional small curvature tunneling. Internal rotation of involved molecules is taken explicitly into account in the molecular partition function. The rate coefficients are calculated in the temperature range 300–900 K, relevant to low-temperature autoignition. The concerted HO2 elimination is an important reaction that competes with some H-transfer and is associated with chain termination. Thus, the branching ratio between these reaction channels is analyzed. We show that variational and multidimensional tunneling effects cannot be neglected for the H-transfer reaction. In particular, the pre-exponential Arrhenius fitting parameter derived from our rate constants shows a strong dependence on the temperature, because tunneling increases quickly at temperatures below 500 K. On the basis of our results, the existing qualitative model for the reasons for different knock behavior observed for n-heptane and isooctane is quantitatively validated at the molecular level.
中文翻译:

评估燃料辛烷值规模的分子基础:庚烷和异辛烷自燃速率控制步骤的详细研究
N-庚烷和2,2,4-三甲基戊烷(异辛烷)是烃类燃料配方点火模型中的关键物质。异辛烷具有抗爆震性,而正庚烷是一种极易爆震的烃。已经提出,通过氢转移异构化反应使它们相关的烷基过氧烷基和氢过氧烷基物种相互转化是理解它们不同的爆震行为的关键步骤。在这项工作中,n的单分子氢转移反应的动力学-正庚基过氧基和异辛基过氧基是使用规范的变分过渡态理论和多维小曲率隧穿确定的。在分子分配功能中明确考虑了所涉及分子的内部旋转。速率系数是在300-900 K的温度范围内计算的,与低温自燃有关。齐心协力的HO 2消除是一个重要的反应,与某些H转移竞争,并与链终止相关。因此,分析了这些反应通道之间的支化率。我们表明,对于H转移反应,不能忽略变构和多维隧穿效应。特别是,从我们的速率常数得出的指数前Arrhenius拟合参数显示出对温度的强烈依赖性,因为在低于500 K的温度下隧穿迅速增加。根据我们的结果,由于不同爆震的原因,现有的定性模型对正庚烷和异辛烷的观察到的行为在分子水平上得到了定量验证。
更新日期:2018-01-05
中文翻译:
评估燃料辛烷值规模的分子基础:庚烷和异辛烷自燃速率控制步骤的详细研究
N-庚烷和2,2,4-三甲基戊烷(异辛烷)是烃类燃料配方点火模型中的关键物质。异辛烷具有抗爆震性,而正庚烷是一种极易爆震的烃。已经提出,通过氢转移异构化反应使它们相关的烷基过氧烷基和氢过氧烷基物种相互转化是理解它们不同的爆震行为的关键步骤。在这项工作中,n的单分子氢转移反应的动力学-正庚基过氧基和异辛基过氧基是使用规范的变分过渡态理论和多维小曲率隧穿确定的。在分子分配功能中明确考虑了所涉及分子的内部旋转。速率系数是在300-900 K的温度范围内计算的,与低温自燃有关。齐心协力的HO 2消除是一个重要的反应,与某些H转移竞争,并与链终止相关。因此,分析了这些反应通道之间的支化率。我们表明,对于H转移反应,不能忽略变构和多维隧穿效应。特别是,从我们的速率常数得出的指数前Arrhenius拟合参数显示出对温度的强烈依赖性,因为在低于500 K的温度下隧穿迅速增加。根据我们的结果,由于不同爆震的原因,现有的定性模型对正庚烷和异辛烷的观察到的行为在分子水平上得到了定量验证。










































 京公网安备 11010802027423号
京公网安备 11010802027423号