当前位置:
X-MOL 学术
›
Acta Mater.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
First-principles study of crystal structure and stability of T 1 precipitates in Al-Li-Cu alloys
Acta Materialia ( IF 8.3 ) Pub Date : 2018-02-01 , DOI: 10.1016/j.actamat.2017.12.013 Kyoungdoc Kim , Bi-Cheng Zhou , C. Wolverton
Acta Materialia ( IF 8.3 ) Pub Date : 2018-02-01 , DOI: 10.1016/j.actamat.2017.12.013 Kyoungdoc Kim , Bi-Cheng Zhou , C. Wolverton
|
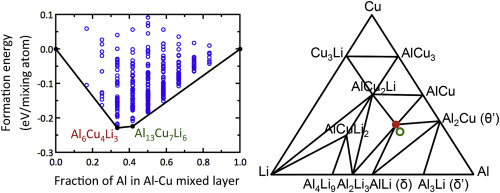
Abstract Aluminum-lithium-copper alloys have a low density, high elastic modulus and high specific strength. Due to this combination of properties, alloys strengthened with the ternary (Al-Li-Cu) T 1 phase have attracted a great deal of interest especially in aerospace applications. Determining the atomic structural information of the precipitate is a fundamental step in developing a basis for advanced alloy design; however, even though many experimental studies have addressed the T 1 crystal structure, it remains the subject of some controversy. Here, we use density functional theory (DFT) calculations to investigate the structure and composition of the T 1 phase by comparing the energetic stability of five previously-proposed models of the crystal structure of T 1 . The DFT formation energy of these proposed T 1 crystal structures was calculated using a special quasi-random structure (SQS) approach to describe a disordered Al-Cu sub-lattice. In conflict with the experimental phase diagram, DFT calculations of all five proposed models result in an energetically unstable T 1 phase. We search for a new, lower-energy structure of T 1 using a cluster expansion approach, and find a new structural model with DFT energy that is stable (at T = 0 K), i.e., on the calculated convex hull of the Al-Li-Cu ternary system. However, this new predicted phase does not have a tie-line with Al, but the formation energy of the phase is very close to the energy required to make a tie-line with Al ( Δ E = 0.013 eV/atom), which could be affected by finite temperature entropic effects (i.e., vibrational entropic stabilization).
中文翻译:

Al-Li-Cu合金中T 1 析出物晶体结构和稳定性的第一性原理研究
摘要 铝锂铜合金具有密度低、弹性模量高和比强度高等特点。由于这种性能的组合,用三元 (Al-Li-Cu) T 1 相强化的合金引起了极大的兴趣,尤其是在航空航天应用中。确定析出物的原子结构信息是开发先进合金设计基础的基本步骤;然而,尽管许多实验研究已经解决了 T 1 晶体结构,但它仍然存在一些争议。在这里,我们使用密度泛函理论 (DFT) 计算通过比较五个先前提出的 T 1 晶体结构模型的能量稳定性来研究 T 1 相的结构和组成。这些提出的 T 1 晶体结构的 DFT 形成能是使用特殊的准随机结构 (SQS) 方法计算的,以描述无序的 Al-Cu 亚晶格。与实验相图相冲突,所有五个提议模型的 DFT 计算都导致能量不稳定的 T 1 相。我们使用簇扩展方法寻找 T 1 的新的低能量结构,并找到具有稳定 DFT 能量的新结构模型(在 T = 0 K 时),即在计算的 Al 凸包上锂铜三元体系。然而,这个新的预测相没有与 Al 的连接线,但该相的形成能非常接近与 Al 形成连接线所需的能量(Δ E = 0.013 eV/atom),这可以受有限温度熵效应(即振动熵稳定)的影响。
更新日期:2018-02-01
中文翻译:
Al-Li-Cu合金中T 1 析出物晶体结构和稳定性的第一性原理研究
摘要 铝锂铜合金具有密度低、弹性模量高和比强度高等特点。由于这种性能的组合,用三元 (Al-Li-Cu) T 1 相强化的合金引起了极大的兴趣,尤其是在航空航天应用中。确定析出物的原子结构信息是开发先进合金设计基础的基本步骤;然而,尽管许多实验研究已经解决了 T 1 晶体结构,但它仍然存在一些争议。在这里,我们使用密度泛函理论 (DFT) 计算通过比较五个先前提出的 T 1 晶体结构模型的能量稳定性来研究 T 1 相的结构和组成。这些提出的 T 1 晶体结构的 DFT 形成能是使用特殊的准随机结构 (SQS) 方法计算的,以描述无序的 Al-Cu 亚晶格。与实验相图相冲突,所有五个提议模型的 DFT 计算都导致能量不稳定的 T 1 相。我们使用簇扩展方法寻找 T 1 的新的低能量结构,并找到具有稳定 DFT 能量的新结构模型(在 T = 0 K 时),即在计算的 Al 凸包上锂铜三元体系。然而,这个新的预测相没有与 Al 的连接线,但该相的形成能非常接近与 Al 形成连接线所需的能量(Δ E = 0.013 eV/atom),这可以受有限温度熵效应(即振动熵稳定)的影响。










































 京公网安备 11010802027423号
京公网安备 11010802027423号