当前位置:
X-MOL 学术
›
Chem. Bio. Drug Des.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Interaction of antivirals with a heptameric bundle model of the p7 protein of hepatitis C virus
Chemical Biology & Drug Design ( IF 3.2 ) Pub Date : 2018-01-23 , DOI: 10.1111/cbdd.13162 Sophie L. Dahl,Monoj Mon Kalita,Wolfgang B. Fischer
Chemical Biology & Drug Design ( IF 3.2 ) Pub Date : 2018-01-23 , DOI: 10.1111/cbdd.13162 Sophie L. Dahl,Monoj Mon Kalita,Wolfgang B. Fischer
|
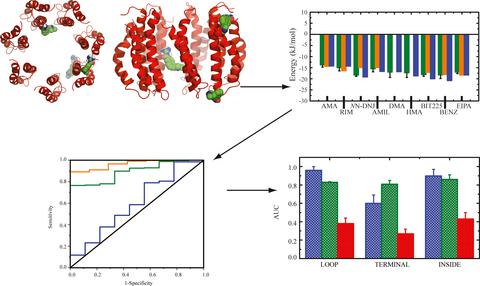
A series of ligands are known experimentally to affect the infectivity cycle of the hepatitis C virus. The target protein for the ligands is proposed to be p7, a 63 amino acid polytopic channel‐forming protein, with possibly two transmembrane domains. Protein p7 is found to assemble into functional oligomers of various sizes, depending on the genotype (GT). Nine ligands are docked to various sites of a computationally derived heptameric bundle of p7 of GT1a. The energy of interaction, here binding energy, is calculated using three different docking programs (Autodock, MOE, LeadIT). Three protein regions are defined to which the ligands are placed, the loop region and the site with the termini as well as the mid‐region which is supposed to track poses inside the putative pore. A common feature is that the loop sites and poses either within the pore or at the intermonomer space of the bundle are preferred for all ligands with proposed binding energies smaller than −10 kJ/mol. BIT225, benzamine, amantadine, and NN‐DNJ show good overall scoring.
中文翻译:

抗病毒药物与丙型肝炎病毒p7蛋白七聚体模型的相互作用
实验上已知一系列配体会影响丙型肝炎病毒的感染周期。配体的目标蛋白被提议为p7,这是一个63个氨基酸的多主题通道形成蛋白,可能具有两个跨膜结构域。发现蛋白p7组装成各种大小的功能性寡聚物,这取决于基因型(GT)。九个配体对接至GT1a p7的计算衍生的七聚体束的各个位点。使用三种不同的对接程序(Autodock,MOE,LeadIT)。定义了三个蛋白质区域,配体被放置在该区域,环区域和带有末端的位点,以及应该追踪假定的孔内姿势的中间区域。一个共同的特征是,对于所有拟议的结合能小于-10 kJ / mol的配体,环的位置和位置都位于束的孔内或单体的单体间空间中。BIT225,苯甲胺,金刚烷胺和N N-DNJ显示出良好的整体评分。
更新日期:2018-01-23
中文翻译:
抗病毒药物与丙型肝炎病毒p7蛋白七聚体模型的相互作用
实验上已知一系列配体会影响丙型肝炎病毒的感染周期。配体的目标蛋白被提议为p7,这是一个63个氨基酸的多主题通道形成蛋白,可能具有两个跨膜结构域。发现蛋白p7组装成各种大小的功能性寡聚物,这取决于基因型(GT)。九个配体对接至GT1a p7的计算衍生的七聚体束的各个位点。使用三种不同的对接程序(Autodock,MOE,LeadIT)。定义了三个蛋白质区域,配体被放置在该区域,环区域和带有末端的位点,以及应该追踪假定的孔内姿势的中间区域。一个共同的特征是,对于所有拟议的结合能小于-10 kJ / mol的配体,环的位置和位置都位于束的孔内或单体的单体间空间中。BIT225,苯甲胺,金刚烷胺和N N-DNJ显示出良好的整体评分。










































 京公网安备 11010802027423号
京公网安备 11010802027423号