当前位置:
X-MOL 学术
›
ACS Cent. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Theoretical Investigations of the Electrochemical Reduction of CO on Single Metal Atoms Embedded in Graphene
ACS Central Science ( IF 12.7 ) Pub Date : 2017-12-18 00:00:00 , DOI: 10.1021/acscentsci.7b00442 Charlotte Kirk 1, 2 , Leanne D. Chen 2, 3 , Samira Siahrostami 1 , Mohammadreza Karamad 1 , Michal Bajdich 2 , Johannes Voss 2 , Jens K. Nørskov 1, 2 , Karen Chan 2
ACS Central Science ( IF 12.7 ) Pub Date : 2017-12-18 00:00:00 , DOI: 10.1021/acscentsci.7b00442 Charlotte Kirk 1, 2 , Leanne D. Chen 2, 3 , Samira Siahrostami 1 , Mohammadreza Karamad 1 , Michal Bajdich 2 , Johannes Voss 2 , Jens K. Nørskov 1, 2 , Karen Chan 2
Affiliation
|
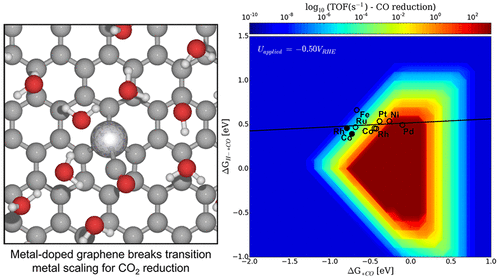
Single transition metal atoms embedded at single vacancies of graphene provide a unique paradigm for catalytic reactions. We present a density functional theory study of such systems for the electrochemical reduction of CO. Theoretical investigations of CO electrochemical reduction are particularly challenging in that electrochemical activation energies are a necessary descriptor of activity. We determined the electrochemical barriers for key proton–electron transfer steps using a state-of-the-art, fully explicit solvent model of the electrochemical interface. The accuracy of GGA-level functionals in describing these systems was also benchmarked against hybrid methods. We find the first proton transfer to form CHO from CO to be a critical step in C1 product formation. On these single atom sites, the corresponding barrier scales more favorably with the CO binding energy than for 211 and 111 transition metal surfaces, in the direction of improved activity. Intermediates and transition states for the hydrogen evolution reaction were found to be less stable than those on transition metals, suggesting a higher selectivity for CO reduction. We present a rate volcano for the production of methane from CO. We identify promising candidates with high activity, stability, and selectivity for the reduction of CO. This work highlights the potential of these systems as improved electrocatalysts over pure transition metals for CO reduction.
中文翻译:

石墨烯中嵌入的单金属原子上CO电化学还原的理论研究
嵌入在石墨烯的单个空位处的单个过渡金属原子为催化反应提供了独特的范例。我们提出了一种用于CO电化学还原的系统的密度泛函理论研究。CO电化学还原的理论研究特别具有挑战性,因为电化学活化能是活性的必要描述。我们使用最先进的,完全明确的电化学界面溶剂模型,确定了关键质子-电子转移步骤的电化学势垒。GGA级功能在描述这些系统时的准确性也以混合方法为基准。我们发现从CO形成CHO的首次质子转移是C 1中的关键步骤产品形成。在这些单原子位点上,与211和111过渡金属表面相比,在提高活性的方向上,与CO结合能相比,相应的势垒更有利地按比例缩放。发现用于氢释放反应的中间体和过渡态的稳定性比过渡金属不稳定,表明对CO还原的选择性更高。我们提出了一种由CO生成甲烷的速率火山。我们确定了具有高活性,稳定性和CO还原选择性的有前途的候选者。这项工作强调了这些系统作为改进的电催化剂优于CO还原纯过渡金属的潜力。
更新日期:2017-12-18
中文翻译:
石墨烯中嵌入的单金属原子上CO电化学还原的理论研究
嵌入在石墨烯的单个空位处的单个过渡金属原子为催化反应提供了独特的范例。我们提出了一种用于CO电化学还原的系统的密度泛函理论研究。CO电化学还原的理论研究特别具有挑战性,因为电化学活化能是活性的必要描述。我们使用最先进的,完全明确的电化学界面溶剂模型,确定了关键质子-电子转移步骤的电化学势垒。GGA级功能在描述这些系统时的准确性也以混合方法为基准。我们发现从CO形成CHO的首次质子转移是C 1中的关键步骤产品形成。在这些单原子位点上,与211和111过渡金属表面相比,在提高活性的方向上,与CO结合能相比,相应的势垒更有利地按比例缩放。发现用于氢释放反应的中间体和过渡态的稳定性比过渡金属不稳定,表明对CO还原的选择性更高。我们提出了一种由CO生成甲烷的速率火山。我们确定了具有高活性,稳定性和CO还原选择性的有前途的候选者。这项工作强调了这些系统作为改进的电催化剂优于CO还原纯过渡金属的潜力。










































 京公网安备 11010802027423号
京公网安备 11010802027423号