当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Structure-Based Kinase Profiling To Understand the Polypharmacological Behavior of Therapeutic Molecules
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2017-12-15 00:00:00 , DOI: 10.1021/acs.jcim.7b00227 Devawati Dutta 1 , Ranjita Das 1 , Chhabinath Mandal 2 , Chitra Mandal 1
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2017-12-15 00:00:00 , DOI: 10.1021/acs.jcim.7b00227 Devawati Dutta 1 , Ranjita Das 1 , Chhabinath Mandal 2 , Chitra Mandal 1
Affiliation
|
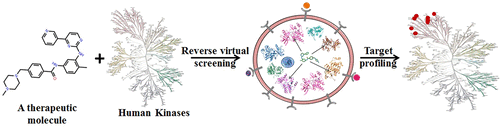
Several drugs elicit their therapeutic efficacy by modulating multiple cellular targets and possess varied polypharmacological actions. The identification of the molecular targets of a potent bioactive molecule is essential in determining its overall polypharmacological profile. Experimental procedures are expensive and time-consuming. Therefore, computational approaches are actively implemented in rational drug discovery. Here, we demonstrate a computational pipeline, based on reverse virtual screening technique using several consensus scoring strategies, and perform structure-based kinase profiling of 12 FDA-approved drugs. This target prediction showed an overall good performance, with an average AU-ROC greater than 0.85 for most drugs, and identified the true targets even at the top 2% cutoff. In contrast, 10 non-kinase binder drugs exhibited lower binding efficiency and appeared in the bottom of ranking list. Subsequently, we validated this pipeline on a potent therapeutic molecule, mahanine, whose polypharmacological profile related to targeting kinases is unknown. Our target-prediction method identified different kinases. Furthermore, we have experimentally validated that mahanine is able to modulate multiple kinases that are involved in cross-talk with different signaling molecules, which thereby exhibits its polypharmacological action. More importantly, in vitro kinase assay exhibited the inhibitory effect of mahanine on two such predicted kinases’ (mTOR and VEGFR2) activity, with IC50 values being ∼12 and ∼22 μM, respectively. Next, we generated a comprehensive drug–protein interaction fingerprint that explained the basis of their target selectivity. We observed that it is controlled by variations in kinase conformations followed by significant differences in crucial hydrogen-bond and van der Waals interactions. Such structure-based kinase profiling could provide useful information in revealing the unknown targets of therapeutic molecules from their polypharmacological behavior and would assist in drug discovery.
中文翻译:

基于结构的激酶分析,以了解治疗分子的多药理行为。
几种药物通过调节多个细胞靶标并具有多种药理作用来引起其治疗效果。确定有效生物活性分子的分子靶标对于确定其整体多药理学特征至关重要。实验过程既昂贵又费时。因此,在合理的药物发现中积极地采用了计算方法。在这里,我们展示了一种基于逆向虚拟筛选技术的计算流程,该技术使用了几种共识评分策略,并对12种FDA批准的药物进行了基于结构的激酶分析。该目标预测显示总体良好的性能,大多数药物的平均AU-ROC均大于0.85,并且即使在最高2%的临界值时也能确定真正的目标。相比之下,10种非激酶结合剂表现出较低的结合效率,并出现在排行榜的底部。随后,我们在强大的治疗分子中验证了这一管线,该蛋白与靶向激酶相关的多药理学特征尚不清楚。我们的目标预测方法可识别出不同的激酶。此外,我们已经在实验上验证了花嘌呤能够调节与不同信号分子发生串扰的多种激酶,从而发挥其多药理作用。更重要的是,此外,我们已经在实验上验证了花嘌呤能够调节与不同信号分子发生串扰的多种激酶,从而发挥其多药理作用。更重要的是,此外,我们已经在实验上验证了花嘌呤能够调节与不同信号分子发生串扰的多种激酶,从而发挥其多药理作用。更重要的是,体外激酶测定法显示,大麦碱对两种这样的预测激酶(mTOR和VEGFR2)活性具有抑制作用,IC 50值分别为〜12和〜22μM。接下来,我们生成了全面的药物-蛋白质相互作用指纹图谱,解释了其靶标选择性的基础。我们观察到它受激酶构象的变化控制,随后在关键的氢键和范德华相互作用中存在显着差异。这种基于结构的激酶谱分析可提供有用的信息,以从治疗分子的多药理行为揭示治疗分子的未知靶点,并有助于药物发现。
更新日期:2017-12-15
中文翻译:
基于结构的激酶分析,以了解治疗分子的多药理行为。
几种药物通过调节多个细胞靶标并具有多种药理作用来引起其治疗效果。确定有效生物活性分子的分子靶标对于确定其整体多药理学特征至关重要。实验过程既昂贵又费时。因此,在合理的药物发现中积极地采用了计算方法。在这里,我们展示了一种基于逆向虚拟筛选技术的计算流程,该技术使用了几种共识评分策略,并对12种FDA批准的药物进行了基于结构的激酶分析。该目标预测显示总体良好的性能,大多数药物的平均AU-ROC均大于0.85,并且即使在最高2%的临界值时也能确定真正的目标。相比之下,10种非激酶结合剂表现出较低的结合效率,并出现在排行榜的底部。随后,我们在强大的治疗分子中验证了这一管线,该蛋白与靶向激酶相关的多药理学特征尚不清楚。我们的目标预测方法可识别出不同的激酶。此外,我们已经在实验上验证了花嘌呤能够调节与不同信号分子发生串扰的多种激酶,从而发挥其多药理作用。更重要的是,此外,我们已经在实验上验证了花嘌呤能够调节与不同信号分子发生串扰的多种激酶,从而发挥其多药理作用。更重要的是,此外,我们已经在实验上验证了花嘌呤能够调节与不同信号分子发生串扰的多种激酶,从而发挥其多药理作用。更重要的是,体外激酶测定法显示,大麦碱对两种这样的预测激酶(mTOR和VEGFR2)活性具有抑制作用,IC 50值分别为〜12和〜22μM。接下来,我们生成了全面的药物-蛋白质相互作用指纹图谱,解释了其靶标选择性的基础。我们观察到它受激酶构象的变化控制,随后在关键的氢键和范德华相互作用中存在显着差异。这种基于结构的激酶谱分析可提供有用的信息,以从治疗分子的多药理行为揭示治疗分子的未知靶点,并有助于药物发现。










































 京公网安备 11010802027423号
京公网安备 11010802027423号