当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Relative Binding Free Energy Calculations in Drug Discovery: Recent Advances and Practical Considerations
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2017-12-15 00:00:00 , DOI: 10.1021/acs.jcim.7b00564 Zoe Cournia 1 , Bryce Allen 2 , Woody Sherman 2
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2017-12-15 00:00:00 , DOI: 10.1021/acs.jcim.7b00564 Zoe Cournia 1 , Bryce Allen 2 , Woody Sherman 2
Affiliation
|
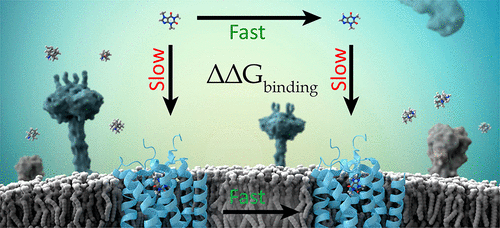
Accurate in silico prediction of protein–ligand binding affinities has been a primary objective of structure-based drug design for decades due to the putative value it would bring to the drug discovery process. However, computational methods have historically failed to deliver value in real-world drug discovery applications due to a variety of scientific, technical, and practical challenges. Recently, a family of approaches commonly referred to as relative binding free energy (RBFE) calculations, which rely on physics-based molecular simulations and statistical mechanics, have shown promise in reliably generating accurate predictions in the context of drug discovery projects. This advance arises from accumulating developments in the underlying scientific methods (decades of research on force fields and sampling algorithms) coupled with vast increases in computational resources (graphics processing units and cloud infrastructures). Mounting evidence from retrospective validation studies, blind challenge predictions, and prospective applications suggests that RBFE simulations can now predict the affinity differences for congeneric ligands with sufficient accuracy and throughput to deliver considerable value in hit-to-lead and lead optimization efforts. Here, we present an overview of current RBFE implementations, highlighting recent advances and remaining challenges, along with examples that emphasize practical considerations for obtaining reliable RBFE results. We focus specifically on relative binding free energies because the calculations are less computationally intensive than absolute binding free energy (ABFE) calculations and map directly onto the hit-to-lead and lead optimization processes, where the prediction of relative binding energies between a reference molecule and new ideas (virtual molecules) can be used to prioritize molecules for synthesis. We describe the critical aspects of running RBFE calculations, from both theoretical and applied perspectives, using a combination of retrospective literature examples and prospective studies from drug discovery projects. This work is intended to provide a contemporary overview of the scientific, technical, and practical issues associated with running relative binding free energy simulations, with a focus on real-world drug discovery applications. We offer guidelines for improving the accuracy of RBFE simulations, especially for challenging cases, and emphasize unresolved issues that could be improved by further research in the field.
中文翻译:

药物发现中的相对结合自由能计算:最新进展和实践考虑
准确的计算机数十年来,基于蛋白质-配体结合亲和力的预测一直是基于结构的药物设计的主要目标,因为它将为药物开发过程带来推定的价值。但是,由于各种科学,技术和实践挑战,计算方法历来未能在现实世界的药物发现应用中提供价值。最近,依赖于基于物理学的分子模拟和统计力学的一系列方法通常被称为相对结合自由能(RBFE)计算,在药物开发项目的背景下可靠地生成准确的预测已显示出希望。这一进步源于基础科学方法(对力场和采样算法的数十年研究)的累积发展,以及计算资源(图形处理单元和云基础架构)的大量增加。回顾性验证研究,盲目的挑战预测和前瞻性应用的大量证据表明,RBFE模拟现在可以以足够的准确性和通量预测同类配体的亲和力差异,从而在潜在客户和潜在客户优化工作中提供可观的价值。在这里,我们概述了当前的RBFE实施方案,重点介绍了最新的进展和尚存的挑战,并举例说明了为获得可靠的RBFE结果而进行实际考虑的示例。我们特别关注相对结合自由能,因为计算量比绝对结合自由能(ABFE)计算要少,并且可以直接映射到“铅对铅”和“铅”最优化过程中,在该过程中可以预测参考分子之间的相对结合能并且可以使用新的想法(虚拟分子)来确定合成分子的优先级。我们结合回顾性文献实例和药物发现项目的前瞻性研究,从理论和应用角度描述了运行RBFE计算的关键方面。这项工作旨在提供与运行相关的结合自由能模拟相关的科学,技术和实践问题的当代概述,重点是现实世界中的药物发现应用。
更新日期:2017-12-15
中文翻译:
药物发现中的相对结合自由能计算:最新进展和实践考虑
准确的计算机数十年来,基于蛋白质-配体结合亲和力的预测一直是基于结构的药物设计的主要目标,因为它将为药物开发过程带来推定的价值。但是,由于各种科学,技术和实践挑战,计算方法历来未能在现实世界的药物发现应用中提供价值。最近,依赖于基于物理学的分子模拟和统计力学的一系列方法通常被称为相对结合自由能(RBFE)计算,在药物开发项目的背景下可靠地生成准确的预测已显示出希望。这一进步源于基础科学方法(对力场和采样算法的数十年研究)的累积发展,以及计算资源(图形处理单元和云基础架构)的大量增加。回顾性验证研究,盲目的挑战预测和前瞻性应用的大量证据表明,RBFE模拟现在可以以足够的准确性和通量预测同类配体的亲和力差异,从而在潜在客户和潜在客户优化工作中提供可观的价值。在这里,我们概述了当前的RBFE实施方案,重点介绍了最新的进展和尚存的挑战,并举例说明了为获得可靠的RBFE结果而进行实际考虑的示例。我们特别关注相对结合自由能,因为计算量比绝对结合自由能(ABFE)计算要少,并且可以直接映射到“铅对铅”和“铅”最优化过程中,在该过程中可以预测参考分子之间的相对结合能并且可以使用新的想法(虚拟分子)来确定合成分子的优先级。我们结合回顾性文献实例和药物发现项目的前瞻性研究,从理论和应用角度描述了运行RBFE计算的关键方面。这项工作旨在提供与运行相关的结合自由能模拟相关的科学,技术和实践问题的当代概述,重点是现实世界中的药物发现应用。










































 京公网安备 11010802027423号
京公网安备 11010802027423号