Cellular Signalling ( IF 4.4 ) Pub Date : 2017-05-07 , DOI: 10.1016/j.cellsig.2017.05.002 Louis M. Luttrell , Stuart Maudsley , Diane Gesty-Palmer
|
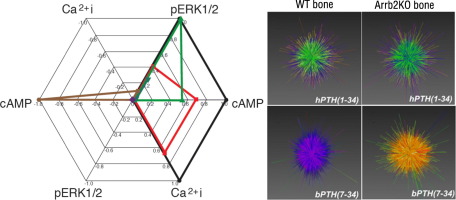
It is increasingly apparent that ligand structure influences both the efficiency with which G protein-coupled receptors (GPCRs) engage their downstream effectors and the manner in which they are activated. Thus, ‘biased’ agonists, synthetic ligands whose intrinsic efficacy differs from the native ligand, afford a strategy for manipulating GPCR signaling in ways that promote beneficial signals while blocking potentially deleterious ones. Still, there are significant challenges in relating in vitro ligand efficacy, which is typically measured in heterologous expression systems, to the biological response in vivo, where the ligand is acting on natively expressed receptors and in the presence of the endogenous ligand. This is particularly true of arrestin pathway-selective ‘biased’ agonists. The type 1 parathyroid hormone receptor (PTH1R) is a case in point. Parathyroid hormone (PTH) is the principal physiological regulator of calcium homeostasis, and PTH1R expressed on cells of the osteoblast lineage are an established therapeutic target in osteoporosis. In vitro, PTH1R signaling is highly sensitive to ligand structure, and PTH analogs that affect the selectivity/kinetics of G protein coupling or that engage arrestin-dependent signaling mechanisms without activating heterotrimeric G proteins have been identified. In vivo, intermittent administration of conventional PTH analogs accelerates the rate of osteoblastic bone formation, largely through known cAMP-dependent mechanisms. Paradoxically, both intermittent and continuous administration of an arrestin pathway-selective PTH analog, which in vivo would be expected to antagonize endogenous PTH1R-cAMP signaling, also increases bone mass. Transcriptomic analysis of tissue from treated animals suggests that conventional and arrestin pathway-selective PTH1R ligands act in largely different ways, with the latter principally affecting pathways involved in the regulation of cell cycle, survival, and migration/cytoskeletal dynamics. Such multi-dimensional in vitro and in vivo analyses of ligand bias may provide insights into the physiological roles of non-canonical arrestin-mediated signaling pathways in vivo, and provide a conceptual framework for translating arrestin pathway-selective ligands into viable therapeutics.
中文翻译:
将体外配体偏倚转化为体内功效
越来越明显的是,配体结构影响着G蛋白偶联受体(GPCR)与其下游效应子结合的效率以及它们被激活的方式。因此,“偏置”激动剂,即内在功效不同于天然配体的合成配体,提供了一种以促进有益信号同时阻断潜在有害信号的方式操纵GPCR信号的策略。然而,将通常在异源表达系统中测量的体外配体功效与体内的生物学反应联系起来仍然存在重大挑战。,其中配体作用于天然表达的受体,并且存在内源性配体。对于抑制蛋白途径选择性的“偏向”激动剂尤其如此。1型甲状旁腺激素受体(PTH 1 R)就是一个例子。甲状旁腺激素(PTH)是钙稳态的主要生理调节剂,成骨细胞谱系细胞上表达的PTH 1 R是骨质疏松症的既定治疗靶标。在体外,PTH 1 R信号传导对配体结构高度敏感,并且已经确定了影响G蛋白偶联的选择性/动力学或参与了抑制蛋白依赖性信号传导机制而不激活异源三聚体G蛋白的PTH类似物。体内常规PTH类似物的间歇给药主要通过已知的cAMP依赖性机制来加速成骨细胞的形成速度。矛盾的是,间歇地和连续地施用抑制素途径选择性的PTH类似物,其在体内可望拮抗内源性PTH 1 R-cAMP信号传导,也增加了骨量。对治疗动物组织进行的转录组分析表明,常规的和抑制蛋白途径选择性的PTH1R配体的作用方式大不相同,后者主要影响参与调节细胞周期,存活和迁移/细胞骨架动力学的途径。例如多维体外和体内配体偏倚的分析可能提供洞察体内非规范性抑制蛋白介导的信号传导途径的生理作用,并为将抑制蛋白途径选择性配体转化为可行的治疗剂提供概念框架。










































 京公网安备 11010802027423号
京公网安备 11010802027423号