当前位置:
X-MOL 学术
›
ACS Earth Space Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Interaction of Elemental Mercury with a Diverse Series of π-Organic Substrates Probed by Computational Methods: Is Mercury Fixation Possible?
ACS Earth and Space Chemistry ( IF 2.9 ) Pub Date : 2017-12-11 00:00:00 , DOI: 10.1021/acsearthspacechem.7b00122 Athanassios C. Tsipis 1
ACS Earth and Space Chemistry ( IF 2.9 ) Pub Date : 2017-12-11 00:00:00 , DOI: 10.1021/acsearthspacechem.7b00122 Athanassios C. Tsipis 1
Affiliation
|
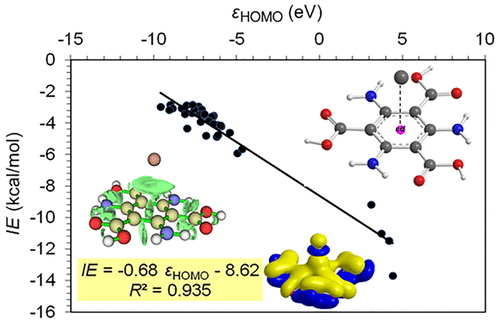
A multitude of electronic structure computational methods has been employed to study the intermolecular adducts formed upon the interaction of elemental mercury, Hg0, with benzene and substituted benzenes of the general formula [Hg(η6-1,3,5-C3H3R3)], where R is either an electron donor substituent, such as −NH2, −OH, −SH, −NH(COCH3), −C≡CH, −S(CH3), −C═CH2, −CH3, −N(CH3)3, and −O(CH3)3 or an electron acceptor substituent, such as −COOH, −NO2, −CHO, −CO(CH3), −CN, −SO2H, −CF3, −C(CO)(NH2), −Cl, −F, and −C(CO)Cl. In addition, a set of neutral and negatively charged octupolar molecules, with alternating electron donor and electron acceptor groups in the 1,3,5 and 2,4,6 positions of the benzene ring, have been included in this study. The estimated interaction energies of Hg0 with these systems range from −2.6 to −13.7 kcal/mol. The interplay of electrostatic, dispersion forces and covalent interaction components constitute the Hg0···π-organic substrate interactions. In the [Hg(η6-1,3,5-C3H3R3)] adducts with electron donor R substituents, the electrostatic forces dominate over the dispersion forces, while the opposite is true for the [Hg(η6-1,3,5-C3H3R3)] adducts with electron acceptor R substituents. In both cases, the covalent component of the interactions is estimated to be marginal. In the interactions of Hg0 with octupolar systems, the electrostatic and dispersion forces are dominant, with the exception of the negatively charged systems, where covalent interactions overwhelm dispersion forces and, along with electrostatic interactions, are the dominant forces. The calculated interaction energies of Hg0 with the π-organic substrates are correlated with the eigenvalues of the highest occupied molecular orbital (HOMO) (εHOMO) and lowest unoccupied molecular orbital (LUMO) (εLUMO) of the substrates as well as their chemical potential (μ). Noteworthy, the orbital interaction components are modulated by εHOMO and εLUMO, while the electrostatic interaction components are modulated by μ. The dispersion force component of the interactions correlates well with the polarizability tensor element in the z axis, αzz. The computations revealed that mercury fixation by π-organic substrates is possible, thus helping to design π-organic linkers in novel MOF sorbents with fast, most efficient, and even reversible Hg0 fixation.
中文翻译:

元素汞与通过计算方法探测的一系列π-有机底物的相互作用:汞固定是否可行?
的电子结构计算方法的大量已被用来研究在元素汞的相互作用,汞形成的分子间的加合物0,用苯和取代的通式[汞苯(η 6 -1,3,5--C 3 ħ 3 R 3)],其中R是电子给体取代基,例如-NH 2,-OH,-SH,-NH(COCH 3),-C≡CH,-S(CH 3),-C═CH 2,-CH 3,-N(CH 3)3和-O(CH 3)3或电子受体取代基,例如-COOH,-NO 2,-CHO,-CO(CH 3),-CN,-SO 2 H,-CF 3,-C(CO)(NH 2),-Cl,-F和-C(CO)Cl。此外,本研究还包括一组中性和带负电荷的八极分子,在苯环的1,3,5和2,4,6位置具有交替的电子供体和电子受体基团。Hg 0与这些系统的估计相互作用能范围为-2.6至-13.7 kcal / mol。静电,分散力和共价相互作用成分之间的相互作用构成了Hg 0 ···π-有机底物相互作用。在[汞柱(η 6 -1,3,5--C 3 H ^ 3 - [R 3)]与电子给体的R取代基的加合物,所述静电力主导在色散力,而相反的是用于[汞(真η 6 -1,3,5--C 3 H ^ 3 - [R 3)]与电子受体- [R加合物取代基。在这两种情况下,相互作用的共价成分估计都是微不足道的。在Hg 0与八极体系的相互作用中,静电力和分散力占主导地位,带负电荷的系统除外,在该体系中,共价相互作用压倒了分散力,并且与静电相互作用一起成为主导力。计算出的Hg 0的相互作用能与π-有机底物的最高占据分子轨道(HOMO)的特征值(ε相关HOMO)和最低未占分子轨道(LUMO)(ε LUMO)的基板的以及它们的化学势(μ)。值得注意的是,轨道相互作用分量由εHOMO和εLUMO调制,而静电相互作用分量由μ调制。相互作用的色散分量与z轴上的极化张量元素αzz很好地相关。计算表明,通过π-有机底物固定汞是可行的,从而有助于在新型MOF吸附剂中以快速,最有效,甚至可逆的Hg 0固定来设计π-有机连接体。
更新日期:2017-12-11
中文翻译:
元素汞与通过计算方法探测的一系列π-有机底物的相互作用:汞固定是否可行?
的电子结构计算方法的大量已被用来研究在元素汞的相互作用,汞形成的分子间的加合物0,用苯和取代的通式[汞苯(η 6 -1,3,5--C 3 ħ 3 R 3)],其中R是电子给体取代基,例如-NH 2,-OH,-SH,-NH(COCH 3),-C≡CH,-S(CH 3),-C═CH 2,-CH 3,-N(CH 3)3和-O(CH 3)3或电子受体取代基,例如-COOH,-NO 2,-CHO,-CO(CH 3),-CN,-SO 2 H,-CF 3,-C(CO)(NH 2),-Cl,-F和-C(CO)Cl。此外,本研究还包括一组中性和带负电荷的八极分子,在苯环的1,3,5和2,4,6位置具有交替的电子供体和电子受体基团。Hg 0与这些系统的估计相互作用能范围为-2.6至-13.7 kcal / mol。静电,分散力和共价相互作用成分之间的相互作用构成了Hg 0 ···π-有机底物相互作用。在[汞柱(η 6 -1,3,5--C 3 H ^ 3 - [R 3)]与电子给体的R取代基的加合物,所述静电力主导在色散力,而相反的是用于[汞(真η 6 -1,3,5--C 3 H ^ 3 - [R 3)]与电子受体- [R加合物取代基。在这两种情况下,相互作用的共价成分估计都是微不足道的。在Hg 0与八极体系的相互作用中,静电力和分散力占主导地位,带负电荷的系统除外,在该体系中,共价相互作用压倒了分散力,并且与静电相互作用一起成为主导力。计算出的Hg 0的相互作用能与π-有机底物的最高占据分子轨道(HOMO)的特征值(ε相关HOMO)和最低未占分子轨道(LUMO)(ε LUMO)的基板的以及它们的化学势(μ)。值得注意的是,轨道相互作用分量由εHOMO和εLUMO调制,而静电相互作用分量由μ调制。相互作用的色散分量与z轴上的极化张量元素αzz很好地相关。计算表明,通过π-有机底物固定汞是可行的,从而有助于在新型MOF吸附剂中以快速,最有效,甚至可逆的Hg 0固定来设计π-有机连接体。










































 京公网安备 11010802027423号
京公网安备 11010802027423号