当前位置:
X-MOL 学术
›
Catal. Sci. Technol.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
DFT study of CO2 hydrogenation catalyzed by a cobalt-based system: an unexpected formate anion-assisted deprotonation mechanism†
Catalysis Science & Technology ( IF 4.4 ) Pub Date : 2017-12-12 00:00:00 , DOI: 10.1039/c7cy02012k Zhihan Zhang 1, 2, 3, 4, 5 , Yinwu Li 1, 2, 3, 4, 5 , Cheng Hou 1, 2, 3, 4, 5 , Cunyuan Zhao 1, 2, 3, 4, 5 , Zhuofeng Ke 1, 2, 3, 4, 5
Catalysis Science & Technology ( IF 4.4 ) Pub Date : 2017-12-12 00:00:00 , DOI: 10.1039/c7cy02012k Zhihan Zhang 1, 2, 3, 4, 5 , Yinwu Li 1, 2, 3, 4, 5 , Cheng Hou 1, 2, 3, 4, 5 , Cunyuan Zhao 1, 2, 3, 4, 5 , Zhuofeng Ke 1, 2, 3, 4, 5
Affiliation
|
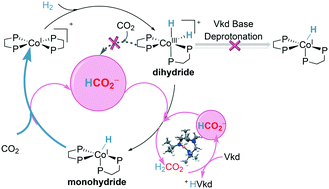
Catalyzed hydrogenation of CO2 by earth-abundant metal complexes is a promising strategy to utilize hydrogen as a kind of sustainable energy and to reduce greenhouse gases. A systematic density functional theory (DFT) study is presented for CO2 hydrogenation catalyzed by the Co(dmpe)2H (dmpe: 1,2-bis(dimethylphosphino)-ethane) complex with Verkade's base as an additive. An unexpected formate anion-assisted deprotonation mechanism is unfolded, which is different from the generally accepted additive base-catalyzed deprotonation mechanism. The complete catalytic cycle involves three main steps: (i) oxidative addition, (ii) deprotonation of the dihydride complex, and (iii) hydrogenation of CO2. The cobalt monohydride complex Co(dmpe)2H is found to be the catalytically active species and the rate-determining step is the hydrogenation of CO2 (ΔG‡ = 20.9 kcal mol−1). Furthermore, the hydride transfer process prefers the reductive elimination mechanism (ΔG‡ = 20.9 kcal mol−1) to the direct transfer mechanism (ΔG‡ = 23.3 kcal mol−1). In contrast, the cobalt dihydride complex [Co(dmpe)2H2]+ is less likely to be the active species, which should be deprotonated to the cobalt monohydride species for further hydrogenation. The deprotonation of cobalt dihydride to cobalt monohydride is found to be promoted by the formate anion (ΔG‡ = 10.5 kcal mol−1) instead of Verkade's base (ΔG‡ = 36.9 kcal mol−1). On the other hand, a direct heterolytic cleavage of H2 assisted by the base to cobalt monohydride is less feasible compared with the oxidative addition of H2. These results can well explain the necessity for oxidative addition and the appearance of formic acid after Verkade's base has run out in the experiments, and are in good agreement with the experimental observation that the hydrogenation of CO2 instead of the deprotonation is the rate-determining step. The present results provide sharp insights and helpful guidelines for designing novel hydrogenation systems with transition metal complexes and bases.
中文翻译:

基于钴的系统催化 的CO 2加氢的DFT研究:意外的甲酸阴离子辅助的去质子化机制†
富含地球的金属络合物催化的CO 2加氢是利用氢作为一种可持续能源并减少温室气体的一种有前途的策略。提出了系统密度泛函理论(DFT)研究,以Verkade碱为添加剂的Co(dmpe)2 H(dmpe:1,2-双(二甲基膦基)-乙烷)配合物催化CO 2加氢。意外的甲酸酯阴离子辅助的去质子化机理得以展开,这与通常公认的添加剂碱催化的去质子化机理不同。完整的催化循环包括三个主要步骤:(i)氧化加成,(ii)二氢配合物的去质子化,和(iii)CO 2的氢化。一氢化钴配合物Co(dmpe)发现2 H是催化活性物质,并且决定速率的步骤是CO 2的氢化( ΔG ‡ = 20.9 kcal mol -1)。此外,氢化物转移过程倾向于还原消除机构(Δ ģ ‡ = 20.9千卡摩尔-1)到直接搬送机构(Δ ģ ‡ = 23.3千卡摩尔-1)。相反,二氢化钴络合物[Co(dmpe) 2 H 2 ] +不太可能是活性物质,应将其脱质子化为一氢化钴物质以进一步氢化。发现由甲酸酯阴离子(ΔG ‡ = 10.5 kcal mol -1)而不是Verkade碱(ΔG ‡ = 36.9 kcal mol -1)促进了二氢化钴向一氢化钴的去质子化。在另一方面,H的直接异裂2与氧化添加H相比由基座协助钴一水合物是不太可行2。这些结果可以很好地说明在实验中用完Verkade碱后氧化加成的必要性和甲酸的出现,并且与实验观察到的一致,即CO 2的氢化而不是去质子化是速率的决定因素。步。目前的结果为设计具有过渡金属配合物和碱的新型氢化系统提供了敏锐的见识和有益的指导。
更新日期:2017-12-12
中文翻译:
基于钴的系统催化 的CO 2加氢的DFT研究:意外的甲酸阴离子辅助的去质子化机制†
富含地球的金属络合物催化的CO 2加氢是利用氢作为一种可持续能源并减少温室气体的一种有前途的策略。提出了系统密度泛函理论(DFT)研究,以Verkade碱为添加剂的Co(dmpe)2 H(dmpe:1,2-双(二甲基膦基)-乙烷)配合物催化CO 2加氢。意外的甲酸酯阴离子辅助的去质子化机理得以展开,这与通常公认的添加剂碱催化的去质子化机理不同。完整的催化循环包括三个主要步骤:(i)氧化加成,(ii)二氢配合物的去质子化,和(iii)CO 2的氢化。一氢化钴配合物Co(dmpe)发现2 H是催化活性物质,并且决定速率的步骤是CO 2的氢化( ΔG ‡ = 20.9 kcal mol -1)。此外,氢化物转移过程倾向于还原消除机构(Δ ģ ‡ = 20.9千卡摩尔-1)到直接搬送机构(Δ ģ ‡ = 23.3千卡摩尔-1)。相反,二氢化钴络合物[Co(dmpe) 2 H 2 ] +不太可能是活性物质,应将其脱质子化为一氢化钴物质以进一步氢化。发现由甲酸酯阴离子(ΔG ‡ = 10.5 kcal mol -1)而不是Verkade碱(ΔG ‡ = 36.9 kcal mol -1)促进了二氢化钴向一氢化钴的去质子化。在另一方面,H的直接异裂2与氧化添加H相比由基座协助钴一水合物是不太可行2。这些结果可以很好地说明在实验中用完Verkade碱后氧化加成的必要性和甲酸的出现,并且与实验观察到的一致,即CO 2的氢化而不是去质子化是速率的决定因素。步。目前的结果为设计具有过渡金属配合物和碱的新型氢化系统提供了敏锐的见识和有益的指导。










































 京公网安备 11010802027423号
京公网安备 11010802027423号