当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Molecular Building Block-Based Electronic Charges for High-Throughput Screening of Metal–Organic Frameworks for Adsorption Applications
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2017-12-29 00:00:00 , DOI: 10.1021/acs.jctc.7b00841 Edwin Argueta 1 , Jeena Shaji 2 , Arun Gopalan 1 , Peilin Liao 3 , Randall Q. Snurr 1 , Diego A. Gómez-Gualdrón 2
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2017-12-29 00:00:00 , DOI: 10.1021/acs.jctc.7b00841 Edwin Argueta 1 , Jeena Shaji 2 , Arun Gopalan 1 , Peilin Liao 3 , Randall Q. Snurr 1 , Diego A. Gómez-Gualdrón 2
Affiliation
|
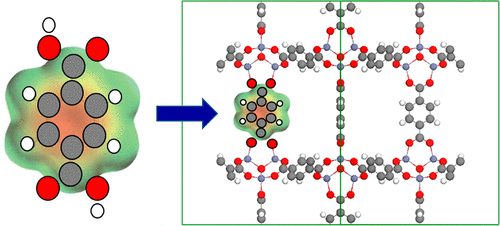
Metal–organic frameworks (MOFs) are porous crystalline materials with attractive properties for gas separation and storage. Their remarkable tunability makes it possible to create millions of MOF variations but creates the need for fast material screening to identify promising structures. Computational high-throughput screening (HTS) is a possible solution, but its usefulness is tied to accurate predictions of MOF adsorption properties. Accurate adsorption simulations often require an accurate description of electrostatic interactions, which depend on the electronic charges of the MOF atoms. HTS-compatible methods to assign charges to MOF atoms need to accurately reproduce electrostatic potentials (ESPs) and be computationally affordable, but current methods present an unsatisfactory trade-off between computational cost and accuracy. We illustrate a method to assign charges to MOF atoms based on ab initio calculations on MOF molecular building blocks. A library of building blocks with built-in charges is thus created and used by an automated MOF construction code to create hundreds of MOFs with charges “inherited” from the constituent building blocks. The molecular building block-based (MBBB) charges are similar to REPEAT charges—which are charges that reproduce ESPs obtained from ab initio calculations on crystallographic unit cells of nanoporous crystals—and thus similar predictions of adsorption loadings, heats of adsorption, and Henry’s constants are obtained with either method. The presented results indicate that the MBBB method to assign charges to MOF atoms is suitable for use in computational high-throughput screening of MOFs for applications that involve adsorption of molecules such as carbon dioxide.
中文翻译:

用于吸附应用的金属-有机骨架的高通量筛选的基于分子构件的电子电荷
金属有机骨架(MOF)是多孔晶体材料,具有吸引人的特性,可用于气体分离和储存。它们出色的可调谐性使其可以创建数百万个MOF变量,但需要快速筛选材料以识别有前途的结构。计算高通量筛选(HTS)是一种可能的解决方案,但其实用性与MOF吸附特性的准确预测有关。准确的吸附模拟通常需要准确描述静电相互作用,这取决于MOF原子的电荷。与HTS兼容的将电荷分配给MOF原子的方法需要准确地复制静电势(ESP),并且在计算上可以承受,但是当前的方法在计算成本和精度之间存在无法令人满意的折衷。我们说明了一种基于对MOF分子构造块的从头算起的将电荷分配给MOF原子的方法。这样就创建了带有内置费用的构建基块库,并由自动化的MOF构造代码使用该库来创建数百个具有从组成构建基块“继承”来的电荷的MOF。基于分子结构单元的电荷(MBBB)与REPEAT电荷类似,后者是重现从纳米多孔晶体的晶体学晶胞从头计算得到的ESP的电荷,因此对吸附负荷,吸附热和亨利常数的预测相似可以通过任何一种方法获得。
更新日期:2017-12-29
中文翻译:
用于吸附应用的金属-有机骨架的高通量筛选的基于分子构件的电子电荷
金属有机骨架(MOF)是多孔晶体材料,具有吸引人的特性,可用于气体分离和储存。它们出色的可调谐性使其可以创建数百万个MOF变量,但需要快速筛选材料以识别有前途的结构。计算高通量筛选(HTS)是一种可能的解决方案,但其实用性与MOF吸附特性的准确预测有关。准确的吸附模拟通常需要准确描述静电相互作用,这取决于MOF原子的电荷。与HTS兼容的将电荷分配给MOF原子的方法需要准确地复制静电势(ESP),并且在计算上可以承受,但是当前的方法在计算成本和精度之间存在无法令人满意的折衷。我们说明了一种基于对MOF分子构造块的从头算起的将电荷分配给MOF原子的方法。这样就创建了带有内置费用的构建基块库,并由自动化的MOF构造代码使用该库来创建数百个具有从组成构建基块“继承”来的电荷的MOF。基于分子结构单元的电荷(MBBB)与REPEAT电荷类似,后者是重现从纳米多孔晶体的晶体学晶胞从头计算得到的ESP的电荷,因此对吸附负荷,吸附热和亨利常数的预测相似可以通过任何一种方法获得。










































 京公网安备 11010802027423号
京公网安备 11010802027423号