当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Machine Learning of Dynamic Electron Correlation Energies from Topological Atoms
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2017-12-06 00:00:00 , DOI: 10.1021/acs.jctc.7b01157 James L. McDonagh 1 , Arnaldo F. Silva 1 , Mark A. Vincent 2 , Paul L. A. Popelier 1, 2
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2017-12-06 00:00:00 , DOI: 10.1021/acs.jctc.7b01157 James L. McDonagh 1 , Arnaldo F. Silva 1 , Mark A. Vincent 2 , Paul L. A. Popelier 1, 2
Affiliation
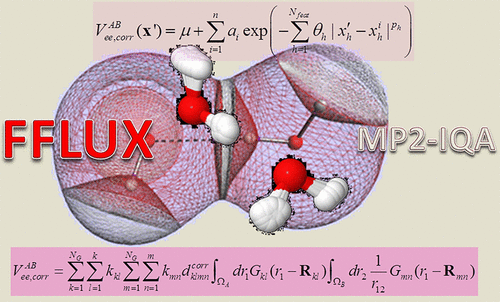
|
We present an innovative method for predicting the dynamic electron correlation energy of an atom or a bond in a molecule utilizing topological atoms. Our approach uses the machine learning method Kriging (Gaussian Process Regression with a non-zero mean function) to predict these dynamic electron correlation energy contributions. The true energy values are calculated by partitioning the MP2 two-particle density-matrix via the Interacting Quantum Atoms (IQA) procedure. To our knowledge, this is the first time such energies have been predicted by a machine learning technique. We present here three important proof-of-concept cases: the water monomer, the water dimer, and the van der Waals complex H2···He. These cases represent the final step toward the design of a full IQA potential for molecular simulation. This final piece will enable us to consider situations in which dispersion is the dominant intermolecular interaction. The results from these examples suggest a new method by which dispersion potentials for molecular simulation can be generated.
中文翻译:

基于拓扑原子的动态电子相关能量的机器学习
我们提出了一种创新的方法,用于预测利用拓扑原子的分子中原子或键的动态电子相关能。我们的方法使用机器学习方法Kriging(具有非零均值函数的高斯过程回归)来预测这些动态电子相关能的贡献。真实能量值是通过交互量子原子(IQA)程序对MP2两粒子密度矩阵进行划分而得出的。据我们所知,这是机器学习技术首次预测这种能量。在这里,我们介绍了三个重要的概念验证案例:水单体,水二聚体和范德华络合物H 2···他。这些情况代表了为分子模拟设计完整IQA潜力的最后一步。这最后一篇文章将使我们能够考虑色散是主要的分子间相互作用的情况。这些示例的结果提出了一种新方法,通过该方法可以生成用于分子模拟的分散电位。
更新日期:2017-12-06
中文翻译:
基于拓扑原子的动态电子相关能量的机器学习
我们提出了一种创新的方法,用于预测利用拓扑原子的分子中原子或键的动态电子相关能。我们的方法使用机器学习方法Kriging(具有非零均值函数的高斯过程回归)来预测这些动态电子相关能的贡献。真实能量值是通过交互量子原子(IQA)程序对MP2两粒子密度矩阵进行划分而得出的。据我们所知,这是机器学习技术首次预测这种能量。在这里,我们介绍了三个重要的概念验证案例:水单体,水二聚体和范德华络合物H 2···他。这些情况代表了为分子模拟设计完整IQA潜力的最后一步。这最后一篇文章将使我们能够考虑色散是主要的分子间相互作用的情况。这些示例的结果提出了一种新方法,通过该方法可以生成用于分子模拟的分散电位。










































 京公网安备 11010802027423号
京公网安备 11010802027423号