当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Scalable Electron Correlation Methods. 5. Parallel Perturbative Triples Correction for Explicitly Correlated Local Coupled Cluster with Pair Natural Orbitals
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2017-12-20 00:00:00 , DOI: 10.1021/acs.jctc.7b01141 Qianli Ma 1 , Hans-Joachim Werner 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2017-12-20 00:00:00 , DOI: 10.1021/acs.jctc.7b01141 Qianli Ma 1 , Hans-Joachim Werner 1
Affiliation
|
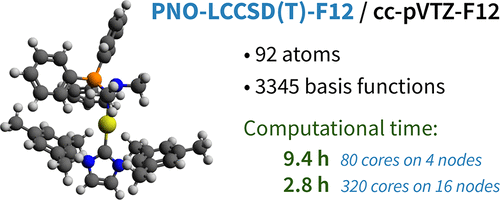
A well-parallelized perturbative triples correction implementation for the pair natural orbital based coupled cluster method PNO-LCCSD(T)-F12 is presented. A composite approach is adopted in addressing the coupling due to off-diagonal Fock matrix elements, in which the local triples amplitudes are iteratively solved using small domains of triples natural orbitals, and a semicanonical (T0) domain correction with larger domains is applied to reduce the domain errors. This treatment adds only about 20% to the computational cost of (T0) calculations with large domains, and the errors due to the use of small domains in the iterations are very small. In addition, a two-step triple list selection method is applied: First, an initial triple list is generated using LCCSD-F12 pair energy criteria, and the (T0) triples energies are computed using small domains. Second, this list is reduced by neglecting triples with small energy contributions, and the final calculation with large domains is only carried out for the reduced list. The cost of the (T) calculation scales asymptotically linear with the molecular size and shows excellent parallelization efficiency up to hundreds of CPU cores. The convergence of the (T) contribution to the relative energies of large molecular systems is carefully tested, and for most of the cases the results obtained with our default thresholds agree within ∼0.5 kcal mol–1 with those computed with very tight thresholds. For all tested molecular systems where canonical calculations are still feasible, the PNO-LCCSD(T)-F12 relative energies also agree within 0.5 kcal mol–1 with the canonical CCSD(T)-F12 results using the same F12 treatment. The (T) calculation generally takes 3–5 times the cost of the preceding PNO-LCCSD-F12 calculation, primarily due to the large number of triples required in obtaining accurate relative energies. We find that for large molecular systems the triple selection criteria used in previous local triples methods are insufficient, and a much larger number of triples are required than it was assumed so far. Still, using a commodity computer cluster, the PNO-LCCSD(T)-F12 calculation of molecules with ∼100 atoms using augmented triple-ζ basis sets can be carried out in a few hours of elapsed time.
中文翻译:

可扩展的电子相关方法。5.具有成对自然轨道的显式相关局部耦合簇的并行摄动三重校正
提出了一种基于偶数自然轨道的耦合簇方法PNO-LCCSD(T)-F12的良好并行的摄动三重校正方法。对于非对角Fock矩阵元素引起的耦合,采用了一种复合方法,其中使用三重自然轨道的小域迭代求解局部三重幅度,并应用具有较大域的半规范(T0)域校正来减少域错误。这种处理仅对使用大域的(T0)计算增加了约20%的计算成本,并且由于在迭代中使用小域而导致的误差非常小。另外,应用了两步三联列表选择方法:首先,使用LCCSD-F12对能量标准生成初始三联列表,(T0)三重态能量是使用小域计算的。其次,通过忽略能量贡献较小的三元组来简化此列表,并且仅对简化的列表执行具有大域的最终计算。(T)计算的成本与分子大小呈渐近线性关系,并在高达数百个CPU内核的情况下显示出出色的并行化效率。(T)对大分子系统相对能量的贡献的收敛性经过仔细测试,在大多数情况下,使用我们的默认阈值获得的结果均在约0.5 kcal mol之内。(T)计算的成本与分子大小呈渐近线性关系,并在高达数百个CPU内核的情况下显示出出色的并行化效率。(T)对大分子系统相对能量的贡献的收敛性经过仔细测试,在大多数情况下,使用我们的默认阈值获得的结果均在约0.5 kcal mol之内。(T)计算的成本与分子大小呈渐近线性关系,并在高达数百个CPU内核的情况下显示出出色的并行化效率。(T)对大分子系统相对能量的贡献的收敛性经过仔细测试,在大多数情况下,使用我们的默认阈值获得的结果均在约0.5 kcal mol之内。–1,并使用非常严格的阈值进行计算。对于仍然可以进行规范计算的所有测试分子系统,PNO-LCCSD(T)-F12的相对能量在0.5 kcal mol –1之内使用相同的F12处理获得规范的CCSD(T)-F12结果。(T)计算通常要花费先前PNO-LCCSD-F12计算成本的3-5倍,这主要是由于获得准确的相对能量需要大量的三元组。我们发现,对于大分子系统,以前的局部三元组方法中使用的三元组选择标准是不够的,并且所需三元组的数量比迄今为止假设的要多得多。仍然,使用商品计算机集群,可以在经过的几个小时内使用增强的Triple-ζ基集对具有约100个原子的分子进行PNO-LCCSD(T)-F12计算。
更新日期:2017-12-20
中文翻译:
可扩展的电子相关方法。5.具有成对自然轨道的显式相关局部耦合簇的并行摄动三重校正
提出了一种基于偶数自然轨道的耦合簇方法PNO-LCCSD(T)-F12的良好并行的摄动三重校正方法。对于非对角Fock矩阵元素引起的耦合,采用了一种复合方法,其中使用三重自然轨道的小域迭代求解局部三重幅度,并应用具有较大域的半规范(T0)域校正来减少域错误。这种处理仅对使用大域的(T0)计算增加了约20%的计算成本,并且由于在迭代中使用小域而导致的误差非常小。另外,应用了两步三联列表选择方法:首先,使用LCCSD-F12对能量标准生成初始三联列表,(T0)三重态能量是使用小域计算的。其次,通过忽略能量贡献较小的三元组来简化此列表,并且仅对简化的列表执行具有大域的最终计算。(T)计算的成本与分子大小呈渐近线性关系,并在高达数百个CPU内核的情况下显示出出色的并行化效率。(T)对大分子系统相对能量的贡献的收敛性经过仔细测试,在大多数情况下,使用我们的默认阈值获得的结果均在约0.5 kcal mol之内。(T)计算的成本与分子大小呈渐近线性关系,并在高达数百个CPU内核的情况下显示出出色的并行化效率。(T)对大分子系统相对能量的贡献的收敛性经过仔细测试,在大多数情况下,使用我们的默认阈值获得的结果均在约0.5 kcal mol之内。(T)计算的成本与分子大小呈渐近线性关系,并在高达数百个CPU内核的情况下显示出出色的并行化效率。(T)对大分子系统相对能量的贡献的收敛性经过仔细测试,在大多数情况下,使用我们的默认阈值获得的结果均在约0.5 kcal mol之内。–1,并使用非常严格的阈值进行计算。对于仍然可以进行规范计算的所有测试分子系统,PNO-LCCSD(T)-F12的相对能量在0.5 kcal mol –1之内使用相同的F12处理获得规范的CCSD(T)-F12结果。(T)计算通常要花费先前PNO-LCCSD-F12计算成本的3-5倍,这主要是由于获得准确的相对能量需要大量的三元组。我们发现,对于大分子系统,以前的局部三元组方法中使用的三元组选择标准是不够的,并且所需三元组的数量比迄今为止假设的要多得多。仍然,使用商品计算机集群,可以在经过的几个小时内使用增强的Triple-ζ基集对具有约100个原子的分子进行PNO-LCCSD(T)-F12计算。










































 京公网安备 11010802027423号
京公网安备 11010802027423号