当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Rigorous pKa Estimation of Amine Species Using Density-Functional Tight-Binding-Based Metadynamics Simulations
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2017-12-21 00:00:00 , DOI: 10.1021/acs.jctc.7b00855 Aditya Wibawa Sakti 1 , Yoshifumi Nishimura 2 , Hiromi Nakai 1, 2, 3, 4
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2017-12-21 00:00:00 , DOI: 10.1021/acs.jctc.7b00855 Aditya Wibawa Sakti 1 , Yoshifumi Nishimura 2 , Hiromi Nakai 1, 2, 3, 4
Affiliation
|
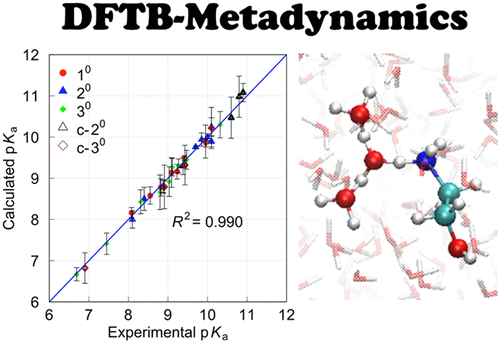
Predicting pKa values for different types of amine species with high accuracy and efficiency is of critical importance for the design of high performance and economical solvents in carbon capture and storage with aqueous amine solutions. In this study, we demonstrate that density-functional tight-binding-based metadynamics simulations are a promising approach to calculate the free energy difference between the protonated and neutral states of amines in aqueous solution with inexpensive computational cost. The calculated pKa values were in satisfactory agreement with the experimental values, the mean absolute deviation being only 0.09 pKa units for 34 amines commonly used in CO2 scrubbing. Such superior reproducibility and correlation compared to estimations by static quantum mechanical calculations highlight the significant effect of dynamical proton transfer processes in explicit solvent molecules for the improvement of the estimation accuracy.
中文翻译:

使用基于密度泛函紧密结合的元动力学模拟对胺物种进行严格的p K a估计
以高精度和高效率预测不同类型胺物种的p K a值,对于在胺水溶液中碳捕集和存储中设计高性能和经济型溶剂至关重要。在这项研究中,我们证明了基于密度泛函紧密结合的元动力学模拟是一种有前途的方法,可以用廉价的计算成本来计算水溶液中胺的质子化和中性状态之间的自由能差。计算出的p K a值与实验值令人满意,平均CO 2中常用的34种胺的绝对绝对偏差仅为0.09 p K a单位擦洗。与通过静态量子力学计算进行的估计相比,这种优越的可重复性和相关性突出了显式溶剂分子中动态质子转移过程对改善估计精度的显著作用。
更新日期:2017-12-21
中文翻译:
使用基于密度泛函紧密结合的元动力学模拟对胺物种进行严格的p K a估计
以高精度和高效率预测不同类型胺物种的p K a值,对于在胺水溶液中碳捕集和存储中设计高性能和经济型溶剂至关重要。在这项研究中,我们证明了基于密度泛函紧密结合的元动力学模拟是一种有前途的方法,可以用廉价的计算成本来计算水溶液中胺的质子化和中性状态之间的自由能差。计算出的p K a值与实验值令人满意,平均CO 2中常用的34种胺的绝对绝对偏差仅为0.09 p K a单位擦洗。与通过静态量子力学计算进行的估计相比,这种优越的可重复性和相关性突出了显式溶剂分子中动态质子转移过程对改善估计精度的显著作用。








































 京公网安备 11010802027423号
京公网安备 11010802027423号